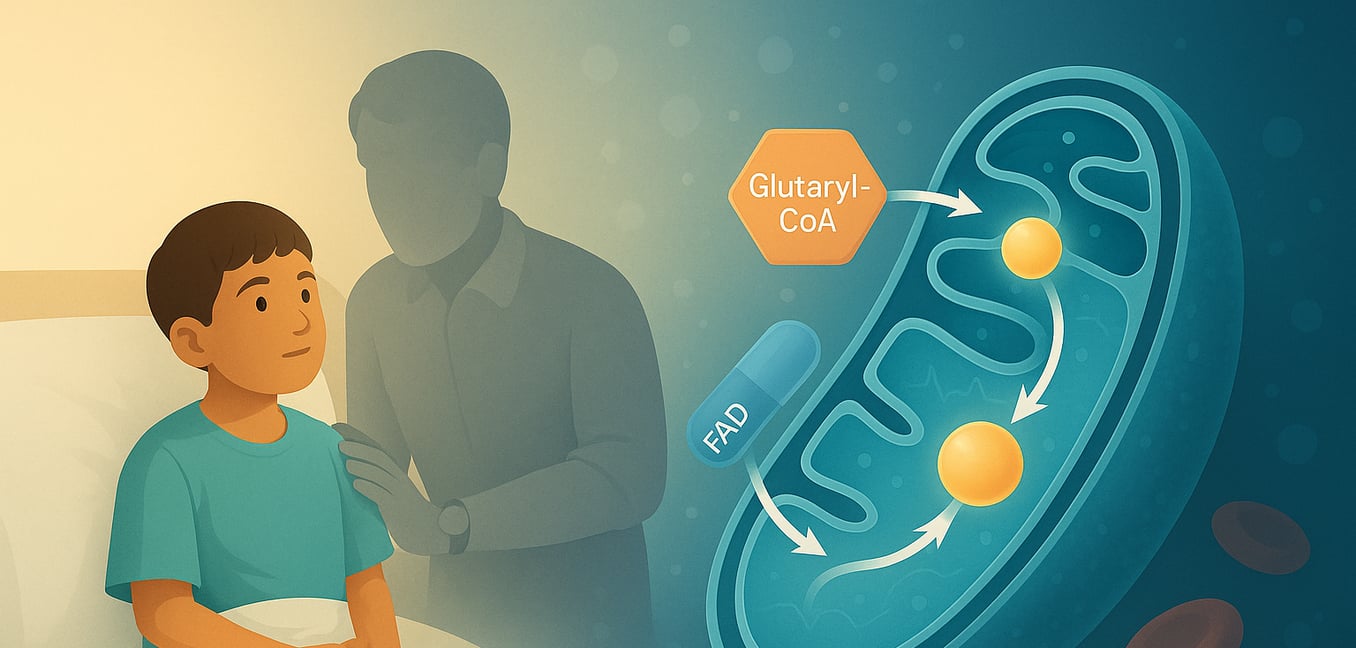



Glutaryl-CoA-Dehydrogenase (GCDH) ist ein wichtiges Enzym, das sich in den Mitochondrien befindet, den Kraftwerken unserer Zellen. Es wird kodiert durch das GCDH Gen, dieses Protein gehört zur Familie der Acyl-CoA-Dehydrogenasen (ACD) und spielt eine entscheidende Rolle im metabolischen Abbau von drei essentiellen Aminosäuren: L-Lysin, L-Hydroxylysin und L-Tryptophan. Die ordnungsgemäße Funktion von GCDH ist entscheidend, um die Ansammlung potenziell schädlicher Substanzen im Körper zu verhindern. Wenn dieses Enzym aufgrund genetischer Mutationen defizitär oder fehlerhaft ist, führt dies zu einer schweren Stoffwechselerkrankung, die als Glutaracidurie Typ I (GA1) bekannt ist, was die Bedeutung seiner Aktivität für die allgemeine Gesundheit, insbesondere die neurologische Entwicklung, unterstreicht.
Die Hauptfunktion von GCDH besteht darin, eine spezifische chemische Reaktion zu katalysieren: die oxidative Decarboxylierung von Glutaryl-CoA zu Crotonyl-CoA und Kohlendioxid. Dieser komplexe Prozess erfolgt in mehreren unterschiedlichen Schritten innerhalb der mitochondrialen Matrix, wo GCDH als Homotetramer existiert (ein Komplex aus vier identischen Untereinheiten). Die Reaktion beginnt, wenn das Substrat Glutaryl-CoA an die oxidierte Form des Enzyms bindet. Ein entscheidender Aminosäurerest in GCDH, Glutamat-370 (Glu370), fungiert als katalytische Base, die ein Proton (das Alpha-Proton) vom Substrat abstrahiert. Danach wird ein Hydridion vom Beta-Kohlenstoff des Substrats an die N(5)-Position des Flavin-Adenin-Dinukleotids (FAD) übertragen, einem Coenzym, das an GCDH gebunden ist. Diese Übertragung reduziert FAD zu FADH2. Dieser Schritt erleichtert die Decarboxylierung (Entfernung einer Carboxylgruppe als CO2) eines enzymgebundenen Intermediats, Glutaconyl-CoA, indem die Cγ-Cδ-Bindung gebrochen wird. Dieser Bruch führt zur Bildung eines Dienolat-Anions, eines Protons und Kohlendioxid. Das Dienolat-Intermediat wird dann protoniert, was das Endprodukt Crotonyl-CoA ergibt, das anschließend zusammen mit CO2 aus dem aktiven Zentrum des Enzyms freigesetzt wird. Um den katalytischen Zyklus abzuschließen und das Enzym für eine weitere Runde wiederherzustellen, wird das reduzierte FADH2 zurück zu FAD oxidiert, indem seine Elektronen in zwei Einzelelektronenschritten an einen externen Elektronenakzeptor übertragen werden, typischerweise an das Elektron-übertragende Flavoprotein (ETF).
Das Verständnis der Funktion von GCDH ist von größter Bedeutung, da dessen Beeinträchtigung schwerwiegende Folgen hat. Bei Menschen mit GA1 führen Mutationen im GCDH Gen zu einem Mangel an funktionellem GCDH-Enzym. Infolgedessen kann der Körper Glutaryl-CoA nicht effektiv verarbeiten. Diese Blockade im Stoffwechselweg führt dazu, dass Glutaryl-CoA und seine vorgelagerten Metaboliten sich ansammeln. Diese werden dann in andere Verbindungen umgewandelt, hauptsächlich in Glutarinsäure (GA), 3-Hydroxyglutarinsäure (3-OH-GA) und in geringerem Maße in Glutaconsäure, die sich in Körperflüssigkeiten und Geweben ansammeln. Diese angesammelten organischen Säuren sind neurotoxin, insbesondere schädlich für bestimmte Bereiche des Gehirns wie die Basalganglien, die entscheidend für die Steuerung von Bewegungen sind. Diese Neurotoxizität ist verantwortlich für die charakteristischen Symptome von GA1, einschließlich akuter encephalopathischer Krisen, Dystonie und anderen schweren Bewegungsstörungen. Die Ansammlung von Glutarylcarnitin (C5DC), einem nicht-toxischen Abfallprodukt, ist ebenfalls ein Merkmal und wird in Neugeborenenscreening-Tests verwendet, um die Erkrankung frühzeitig zu erkennen, um zeitliche Interventionen zu ermöglichen, um die Erkrankung zu managen, zum Beispiel durch Einschränkung der diätetischen Lysinaufnahme.
Was ist ein Mangel an langkettiger Hydroxyacyl-CoA-Dehydrogenase?
Ein Mangel an langkettiger Hydroxyacyl-CoA-Dehydrogenase (LCHAD) ist eine vererbbare Stoffwechselstörung, die verhindert, dass der Körper bestimmte Arten von Fetten, bekannt als langkettige Fettsäuren, effektiv abbaut, um sie in Energie umzuwandeln. Diese Erkrankung tritt aufgrund eines Mangels oder einer Fehlfunktion des LCHAD-Enzyms auf, das eine entscheidende Rolle im mitochondrialen Fettsäure-Beta-Oxidationsweg spielt. Infolgedessen können sich diese unverarbeiteten langkettigen Fettsäuren im Körper ansammeln, was zu potenziell ernsthaften Gesundheitsproblemen führt, die die Leber, das Herz, die Muskeln und das Gehirn betreffen, insbesondere während Perioden des Fastens oder der Krankheit. Zu den wichtigsten Symptomen können niedriger Blutzucker (Hypoglykämie), Muskelschwäche (Myopathie) und Herzprobleme (Kardiomyopathie) gehören, was die Notwendigkeit einer frühen Diagnose und sorgfältigen Behandlung hervorhebt, die oft eine spezielle Diät umfasst.
Was ist die Funktion des PKLR-Gens?
Das PKLR-Gen liefert die wesentlichen Anweisungen zur Synthese des Enzyms Pyruvatkinase, das eine entscheidende Rolle in der zellulären Energieproduktion spielt. Dieses Enzym ist ein Schlüsselspieler in der Glykolyse, dem Stoffwechselweg, der Glucose (eine Art Zucker) in Pyruvat abbaut und dabei Energie freisetzt. Das PKLR-Gen leitet speziell die Produktion von zwei Formen der Pyruvatkinase: dem L-Typ, der überwiegend in der Leber aktiv ist, und dem R-Typ, der in roten Blutkörperchen funktioniert. In beiden Geweben katalysiert die Pyruvatkinase den letzten, energieerzeugenden Schritt der Glykolyse, indem sie die Übertragung einer Phosphatgruppe von Phosphoenolpyruvat auf ADP zur Bildung von ATP, der primären Energiewährung der Zelle, erleichtert.
