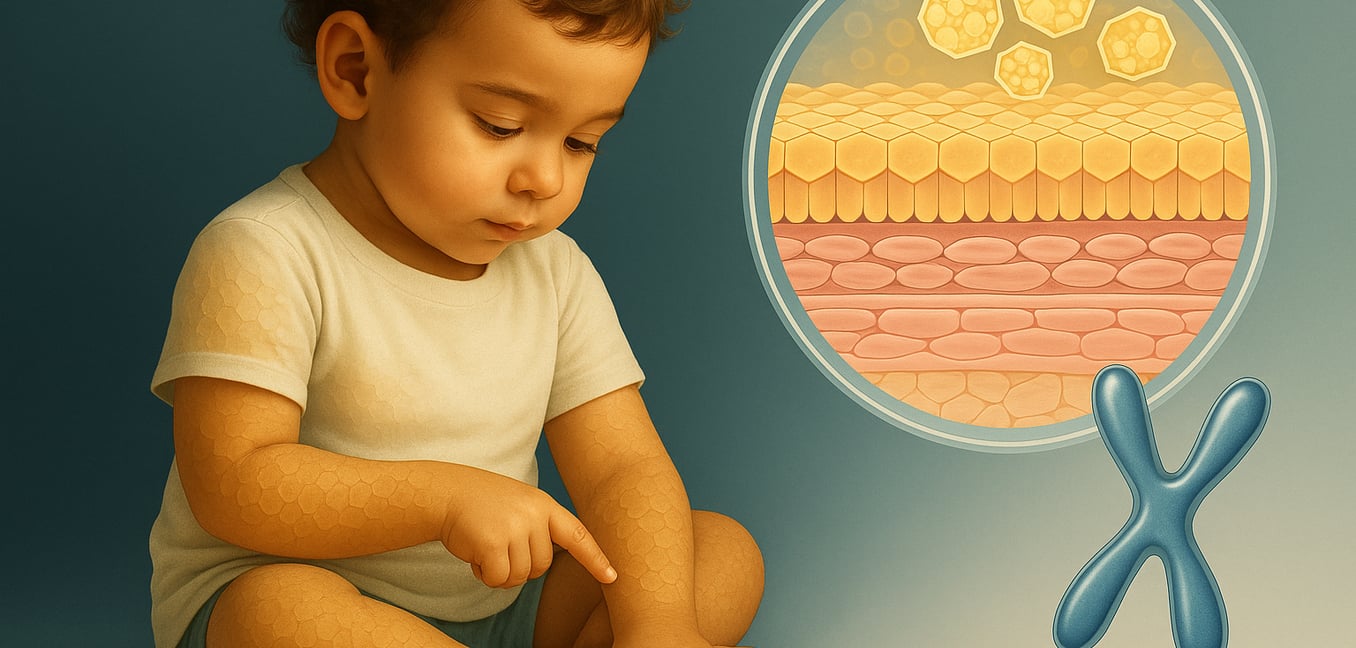



Was ist rezessive X-chromosomale Ichthyose?
Rezessive X-chromosomale Ichthyose (XLRI) ist die zweithäufigste erbliche Form der Ichthyose, einer Familie von Hauterkrankungen, die für trockene, schuppige Haut bekannt sind. Die Erkrankung betrifft fast ausschließlich Männer aufgrund ihres X-chromosomalen Erbgangs. Die Anzeichen treten typischerweise im ersten Lebensjahr auf und zeigen sich als bräunliche, plattenartige Schuppen, die am ausgeprägtesten am Rumpf, am Hals und an den Gliedmaßen zu sehen sind.
Ein wichtiges diagnostisches Merkmal ist das Ausbleiben von Hautfalten, was bedeutet, dass die Falten der Ellenbogen und Knie oft klar bleiben. Die Symptome neigen dazu, sich in den kalten, trockenen Monaten des Winters zu verschlimmern und sich mit der Luftfeuchtigkeit und dem Sonnenlicht im Sommer zu verbessern. Während die Hauterkrankung das Hauptmerkmal ist, kann XLRI auch mit anderen gesundheitlichen Befunden verbunden sein, wie harmlosen trüben Stellen auf der Hornhaut und einem erhöhten Risiko für nicht abgestiegene Hoden. Die Erkrankung wird durch einen Mangel an einem Enzym namens Steroid-Sulfatase verursacht, das den natürlichen Abblätterungsprozess der Haut stört.
Die genetischen Ursachen von XLRI: Ein fehlerhaftes STS-Gen
Die Grundursache der X-chromosomalen Ichthyose ist ein Defekt im Steroid-Sulfatase (STS) Gen, das sich am kurzen Arm des X-Chromosoms befindet. Dieses Gen liefert die Anweisungen zur Produktion des Enzyms Steroid-Sulfatase, eines Proteins, das für die Hautgesundheit essenziell ist. Wenn das STS-Gen fehlerhaft ist, kann der Körper kein funktionales Enzym herstellen, was direkt zu den Symptomen von XLRI führt. Die dafür verantwortlichen genetischen Fehler fallen in zwei Hauptkategorien.
Komplettgenlöschung: Die Hauptursache
In der überwiegenden Mehrheit der Fälle – bis zu 90 % – wird XLRI durch eine vollständige Löschung des STS-Gens verursacht. In diesem Szenario fehlt der gesamte genetische Bauplan des Enzyms auf dem X-Chromosom. Dieser Typ von großflächigen Fehlern tritt oft während der Bildung von Spermien- oder Eizellen auf. Da die Zelle keine Anweisungen hat, kann sie kein Steroid-Sulfatase-Enzym produzieren, was zu den klassischen Merkmalen der Erkrankung führt. Diese große strukturelle Veränderung kann typischerweise mit genetischen Tests nachgewiesen werden, die nach fehlenden Segmenten von Chromosomen suchen, wie z. B. einem chromosomalen Mikroarray.
Punktmutationen: Ein kleiner Fehler mit großer Auswirkung
Bei den verbleibenden 10 % der Personen ist das STS-Gen vorhanden, enthält jedoch einen kleinen, kritischen Fehler, der als Punktmutation bezeichnet wird. Dies ist wie ein einzelner Tippfehler im Code des Gens, der die Anweisungen unbrauchbar macht. Diese Mutationen können entweder ein entscheidendes Element des Enzyms verändern oder ein vorzeitiges "Stopp"-Signal einfügen, das seine Produktion stoppt. Obwohl das Gen nicht vollständig fehlt, sind diese subtilen Veränderungen ausreichend, um ein nicht funktionales Enzym zu erzeugen, was zu demselben klinischen Ergebnis wie bei einer vollständigen Löschung führt. Die Identifizierung dieser kleineren Fehler erfordert eine detailliertere genetische Analyse, wie z. B. DNA-Sequenzierung.
Die biochemischen Auswirkungen: Wie Enzymdefizienz die Haut beeinflusst
Das Steroid-Sulfatase-Enzym ist verantwortlich für den Abbau einer wachsartigen Substanz namens Cholesterinsulfat. Bei XLRI führt das Fehlen dieses Enzyms zu einer Kaskade biochemischer Veränderungen in der Epidermis, der äußersten Hautschicht.
Ansammlung von Cholesterinsulfat
Die primäre Folge des Fehlens des STS-Enzyms ist die Ansammlung von Cholesterinsulfat in der Hornschicht, der obersten Schicht der Haut. In gesunder Haut hält das Enzym diese Werte in Schach. Bei XLRI können die Konzentrationen zehn bis zwanzig Mal höher als normal werden. Diese Ansammlung ist die direkte Ursache für die Hautschuppung und kann in einem Bluttest gemessen werden, um eine Diagnose zu bestätigen.
Versagen der natürlichen Hautabblätterung
Diese Ansammlung verursacht eine "Retention-Hyperkeratose" – ein Begriff für die Unfähigkeit der Haut, ihre toten Zellen abzustoßen. Der natürliche Abblätterungsprozess, genannt Desquamation, beruht auf anderen Enzymen, um das proteinbasierte "Kleber" (Corneodesmosomen) aufzulösen, das alte Hautzellen zusammenhält. Forschungen zeigen, dass überschüssiges Cholesterinsulfat als starkes Inhibitor wirkt, das diese Enzyme im Wesentlichen inaktiviert. Infolgedessen bleibt der Kleber erhalten, und die toten Hautzellen bleiben an Ort und Stelle haften, wodurch die dicken, haftenden Schuppen charakteristisch für XLRI entstehen.
Störung der Hautbarriere
Die schützende Hautbarriere, die Wasserverlust verhindert und Reizstoffe blockiert, hängt von einer hochorganisierten Struktur von Lipiden ab. Die abnormale Ansammlung von Cholesterinsulfat stört dieses empfindliche Gleichgewicht und beeinträchtigt die Integrität der Barriere. Dies trägt nicht nur zur chronischen Trockenheit der Haut bei, sondern verändert auch ihre Gesamtfunktion und Durchlässigkeit.
Komplexe Fälle: Benachbarte Genlöschungs-Syndrome
Während die meisten genetischen Löschungen auf das STS-Gen beschränkt sind, sind einige viel größer und entfernen ein ganzes Viertel benachbarter Gene auf dem X-Chromosom. Wenn dies geschieht, führt es zu einem "benachbarten Genlöschungs-Syndrom", bei dem eine Person XLRI zusammen mit anderen ausgeprägten medizinischen Zuständen hat. Die spezifischen Symptome hängen vollständig davon ab, welche benachbarten Gene ebenfalls fehlen.
Begleitende Erkrankungen bei syndromaler XLRI
Die Löschung von Genen in der Nähe von STS kann zu einem komplexeren klinischen Bild führen. Häufige Beispiele sind:
- Kleinwuchs: Verursacht durch die Löschung des SHOX-Gens, das für die normale Knochenentwicklung und das Wachstum essentiell ist. Dies kann zu einer Erkrankung namens Léri-Weill-Dyschondrosteose führen.
- Kallmann-Syndrom: Resultiert aus der Löschung des KAL1-Gens, was zu einem verminderten oder fehlenden Geruchssinn (Anosmie) und einer verzögerten oder fehlenden Pubertät führt.
- Neuroentwicklungsstörungen: Verbunden mit der Löschung von Genen wie NLGN4X, was das Risiko für geistige Behinderung, Lernschwierigkeiten oder Aufmerksamkeitsdefizit-Hyperaktivitätsstörung (ADHS) erhöhen kann.
