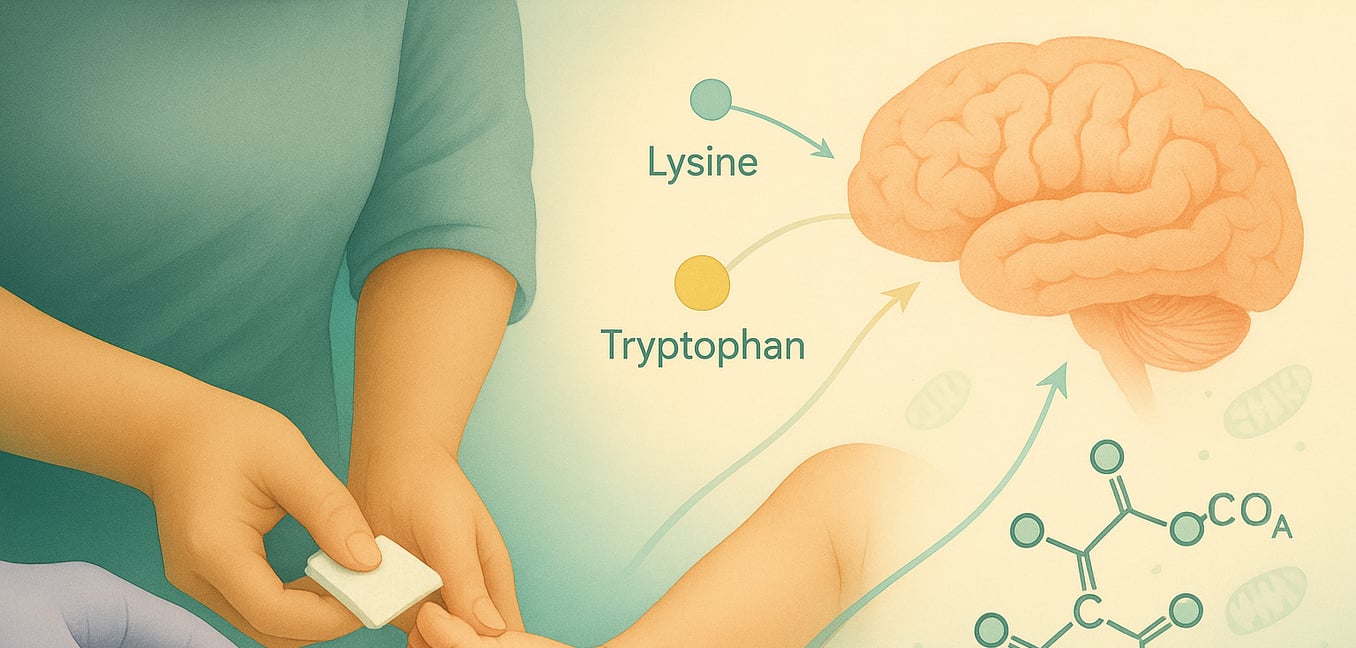



Glutaryl-CoA-Dehydrogenasemangel (GA1), auch bekannt als glutarische Acidämie Typ 1, ist eine erbliche Stoffwechselerkrankung. Sie entsteht aus einem Mangel an dem Enzym Glutaryl-CoA-Dehydrogenase (GCDH), das entscheidend für den Abbau der Aminosäuren Lysin, Hydroxylysin und Tryptophan ist. Dieser enzymatische Defekt führt zur schädlichen Ansammlung von Substanzen wie glutarischer Säure und 3-Hydroxyglutarinsäure, die besonders schädlich für das Gehirn sind, insbesondere für die Basalganglien. GA1 ist eine autosomal-rezessive Erkrankung, was bedeutet, dass ein betroffener Kind von jedem Elternteil ein nicht funktionierendes Exemplar des GCDH-Gens erbt. Das Verständnis des diagnostischen Weges ist entscheidend für eine frühzeitige Intervention und Behandlung.
Neugeborenenscreening: Die erste Linie der Entdeckung
Neugeborenenscreening-Programme spielen eine entscheidende Rolle bei der frühen Identifizierung von GA1, oft bevor Symptome auftreten. Diese frühzeitige Erkennung ist entscheidend, um schwerwiegende neurologische Schäden zu verhindern.
- Screening-Prozess: Eine kleine Blutprobe, die innerhalb von Tagen nach der Geburt über einen Fersenstich entnommen wird, wird analysiert. Labors testen diese getrockneten Blutproben auf erhöhte C5-DC-Acylcarnitinwerte (glutarylcarnitin), ein primärer Marker, der auf potenzielles GA1 hindeutet.
- Ergebnisse interpretieren: Ein Screening-Ergebnis außerhalb des Normalbereichs ist keine definitive Diagnose. Es signalisiert die Notwendigkeit weiterer, spezifischer Bestätigungstests, um festzustellen, ob GA1 vorliegt.
- Bedeutung der frühen Erkennung: Die Identifizierung von GA1 durch Neugeborenenscreening ermöglicht den sofortigen Beginn von Managementstrategien. Dazu gehört eine spezielle, lisinarme Diät und Carnitinsupplementierung, die das Risiko von encephalopathischen Krisen und langfristigen neurologischen Schäden erheblich verringert.
Biochemische Analysen: Bestätigung der Diagnose
Nach einem abnormalen Neugeborenenscreening oder wenn GA1 aufgrund klinischer Symptome vermutet wird, werden spezifische biochemische Tests durchgeführt, um die Diagnose durch die Suche nach charakteristischen metabolischen Signaturen zu bestätigen.
- Urin-Organische-Säure-Analyse: Dieser Test misst die Konzentrationen organischer Säuren im Urin. Bei GA1 sind signifikant erhöhte Konzentrationen von glutarischer Säure und 3-Hydroxyglutarinsäure charakteristische Befunde, die aus dem blockierten Stoffwechselweg resultieren.
- Plasma-Acylcarnitin-Analyse: Eine Blutplasmaprobe wird auf Acylcarnitine analysiert. Erhöhtes Glutarylcarnitin (C5DC) wird typischerweise beobachtet. C5DC-Werte können jedoch manchmal normal oder nur leicht erhöht sein, wenn die Carnitinspeicher des Körpers erschöpft sind, wodurch die Urinanalyse besonders wichtig wird.
- Direkte GCDH-Enzymaktivitätsmessung: Dieser Test bewertet direkt die Funktion des GCDH-Enzyms. Er wird normalerweise an kultivierten Hautzellen (Fibroblasten) oder weißen Blutkörperchen (Leukozyten) durchgeführt. Deutlich reduzierte oder fehlende Enzymaktivität liefert direkte biochemische Beweise für GA1.
Molekulare genetische Tests und enzymatische Assays: definitive Bestätigung
Molekulare genetische Tests und enzymatische Assays bieten die definitiven Bestätigungen von GA1, indem sie den zugrunde liegenden genetischen Defekt und dessen direkten Einfluss auf die Enzymfunktion untersuchen.
- GCDH-Gen-Analyse: Dies umfasst das Sequenzieren des GCDH-Gens zur Identifizierung krankheitsverursachender Varianten (Mutationen). Über 200 solcher Varianten sind bekannt. Die Identifizierung von zwei pathogenen Varianten, eine von jedem Elternteil, bestätigt die Diagnose auf molekularer Ebene.
- Enzymaktivitäts-Tests: Wie in biochemischen Analysen erwähnt, messen diese Tests direkt die Fähigkeit des GCDH-Enzyms, sein Substrat in patientenabgeleiteten Zellen zu verarbeiten. Eine signifikante Reduktion der Aktivität bestätigt den metabolischen Defekt. Während der Rest der Enzymaktivität variieren kann, korreliert sie nicht immer direkt mit der klinischen Schwere.
Neuroimaging-Befunde bei GA1
Neuroimaging-Techniken, hauptsächlich die Magnetresonanztomographie (MRT), sind wertvoll zur Bewertung der Hirnbeteiligung, insbesondere wenn GA1 spät oder nach einer encephalopathischen Krise diagnostiziert wird. Während nicht als primäres diagnostisches Werkzeug zur anfänglichen Identifikation dienen, kann die MRT charakteristische Veränderungen aufzeigen. Diese umfassen häufig:
- Frontotemporale Atrophie (Schrumpfung des Gehirngewebes in den frontalen und temporalen Lappen).
- Erweiterung der Sylvischen Fissuren, manchmal als „Fledermausflügel“-Aussehen beschrieben, aufgrund unvollständiger Operkularisierung.
- Anomalien in den Basalganglien, wie erhöhte Signalintensität auf T2-gewichteten Bildern, die auf Schäden durch Metabolitenansammlungen hinweisen.Diese Befunde helfen, das Ausmaß der neurologischen Auswirkungen zu verstehen und können die Diagnose in Verbindung mit biochemischen und genetischen Ergebnissen unterstützen.
Pränatale Diagnose und genetische Beratung
Für Familien mit einem zuvor betroffenen Kind und bekannten GCDH-Genmutationen bietet die pränatale Diagnostik Optionen für zukünftige Schwangerschaften. Genetische Beratung ist in diesen Fällen entscheidend.
- Zweck: Die pränatale Diagnose zielt darauf ab, festzustellen, ob ein sich entwickelnder Fötus GA1 hat, sodass die Eltern informierte Entscheidungen treffen und sich auf eine spezialisierte Betreuung von der Geburt an vorbereiten können, falls erforderlich.
- Genetische pränatale Tests: Dies umfasst die Analyse von fötalem DNA, die durch Chorionzottenentnahme (CVS) etwa in der 10.-13. Schwangerschaftswoche oder Amniozentese etwa in der 15.-20. Schwangerschaftswoche gewonnen wird. Die fötale DNA wird auf die spezifischen GCDH-Mutationen getestet, die in der Familie identifiziert wurden.
- Biochemische pränatale Tests: Die Konzentrationen von Metaboliten, wie glutarischer Säure, 3-Hydroxyglutarinsäure und Glutarylcarnitin, können in Fruchtwasser, das durch Amniozentese gewonnen wurde, gemessen werden. Erhöhte Werte deuten darauf hin, dass der Fetus betroffen ist, und können die genetischen Tests ergänzen.
- Genetische Beratung: Bietet Familien Informationen über GA1, Vererbungsmuster, Rückfallrisiken und verfügbare Testmöglichkeiten. Sie unterstützt auch Trägertests für andere Familienmitglieder, um deren Risiko zu verstehen, Kinder mit GA1 zu bekommen.
