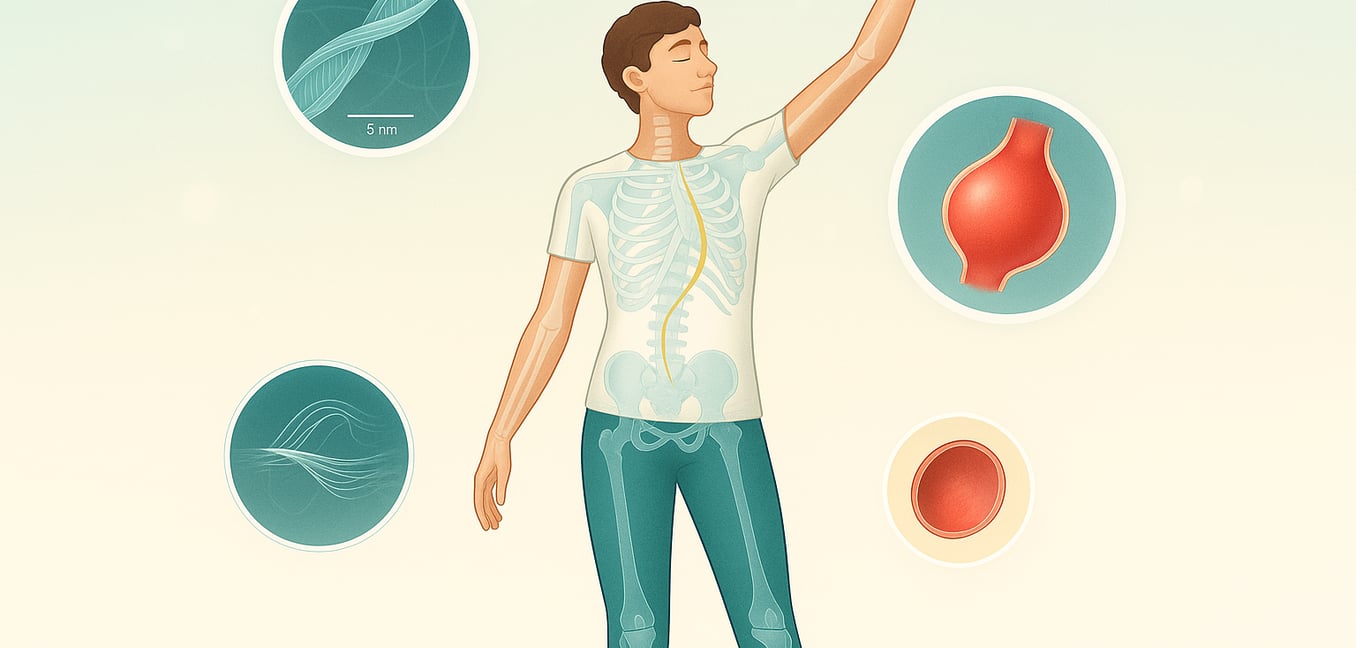



Das Marfan-Syndrom ist eine genetische Erkrankung, die das Bindegewebe des Körpers betrifft, das starke, flexible Material, das Struktur und Unterstützung für Zellen im gesamten Körper bietet. Verursacht durch Varianten im FBN1-Gen, das Anweisungen zur Herstellung eines Proteins namens Fibrillin-1 gibt, schwächt dieser Zustand das Gewebe, das reich an diesem Protein ist. Da Bindegewebe eine grundlegende Komponente vieler verschiedener Organsysteme ist, kann die Erkrankung ein breites Spektrum an gesundheitlichen Problemen verursachen. Das Herz, die Blutgefäße, das Skelett und die Augen sind am häufigsten betroffen, aber die spezifischen Symptome und deren Schwere können von Person zu Person dramatisch variieren, was die Diagnose und das Management zu einem hochgradig individualisierten Prozess macht.
Die Auswirkungen des Marfan-Syndroms sind oft am deutlichsten im Skelettsystem zu erkennen, was zu einem charakteristischen Körperbau und spezifischen knochenbezogenen Problemen führt. Eines der klassischen Anzeichen ist eine große, schlanke Körperform mit überproportional langen Armen, Beinen und Fingern, ein Zustand, der als Arachnodaktylie oder "spinnenartige" Finger bekannt ist. Dies ist auf das Übermaß an langen Knochen zurückzuführen, und betroffene Personen können auch wenig Muskeltonus (Hypotonie) und minimale Fettansammlungen unter der Haut haben. Ein weiteres häufiges Skelettsymptom ist eine Deformität der Brustwand, bei der das Übermaß an Rippen das Brustbein (Sternum) entweder nach innen drückt, um ein eingesunkenes Aussehen zu erzeugen (Pectus excavatum), oder nach außen, was zu einer vorstehenden Brust (Pectus carinatum) führt. Eine Krümmung der Wirbelsäule, bekannt als Skoliose, ist ebenfalls häufig zu beobachten. Diese seitliche Krümmung kann von mild bis schwer variieren und kann während des Wachstums in der Adoleszenz schnell schlimmer werden, was manchmal zu Rückenschmerzen führt. Diese skelettalen Merkmale, zusammen mit anderen wie ungewöhnlich flexiblen Gelenken (Hypermobilität) und Plattfüßen, sind wichtige Indikatoren, nach denen ein Arzt während einer körperlichen Untersuchung sucht.
Über die sichtbaren skelettalen Anzeichen hinaus zeigt das Marfan-Syndrom ernsthafte und potenziell lebensbedrohliche Symptome, die das Herz-Kreislauf-System betreffen. Ein kritisches Anliegen ist die progressive Erweiterung oder Aneurysma der Aorta - der Hauptarterie, die Blut vom Herzen transportiert. Geschwächtes Bindegewebe in der Aortenwand macht es anfällig für Dehnung, und ohne Behandlung kann es reißen (Aortendissektion) oder platzen, was einen medizinischen Notfall darstellt. Diese Schwächung kann auch zu einer Aorteninsuffizienz führen, bei der das Blut zurück ins Herz fließt. Ein weiteres häufiges Herzproblem ist der Mitralklappenprolaps, bei dem die Klappe zwischen den linken Herzkammern schlaff wird und sich nicht richtig schließt. Dies kann zu einer Mitralinsuffizienz führen und in schweren Fällen Brustschmerzen, abnormale Herzrhythmen oder kongestive Herzinsuffizienz zur Folge haben. In den Augen ist ein charakteristisches Symptom die Luxation einer oder beider Linsen (Ektopie lentis), die bei etwa 60 Prozent der Betroffenen auftritt. Diese Verschiebung, zusammen mit starker Kurzsichtigkeit (Myopie), einem erhöhten Risiko für Netzhautablösungen, früh einsetzenden Katarakten und Glaukom, kann zu signifikantem Sehverlust führen, wenn sie nicht sorgfältig von einem Augenfacharzt überwacht und behandelt wird.
Wie sehen Personen mit Marfan aus?
Personen mit Marfan-Syndrom haben oft ein auffälliges physisches Erscheinungsbild, das durch eine große, schlanke Statur mit überproportional langen Armen, Beinen, Fingern (Arachnodaktylie) und Zehen gekennzeichnet ist. Ihre Skelettstruktur kann spezifische Anzeichen zeigen, wie eine Brust, die eingesunken erscheint (Pectus excavatum) oder nach außen vorsteht (Pectus carinatum), sowie eine abnormale seitliche Krümmung der Wirbelsäule (Skoliose). Auffällige Gesichtszüge können ein langes, schmales Gesicht, tief sitzende Augen, einen kleinen Kiefer, flache Wangenknochen und eng stehende Zähne umfassen. Es ist jedoch wichtig zu beachten, dass die physischen Anzeichen bei Einzelpersonen stark variieren und nicht jeder mit dieser Erkrankung all diese Merkmale aufweisen oder sie im gleichen Ausmaß haben wird.
Welche Krankheiten ähneln dem Marfan-Syndrom?
Mehrere vererbbare Bindegewebserkrankungen können dem Marfan-Syndrom (MFS) ähneln, da es signifikante klinische Überlappungen in kardiovaskulären, skelettalen und kutanen Merkmalen gibt. Das Loeys-Dietz-Syndrom (LDS) ist ein Hauptbeispiel, das Merkmale wie Aneurysmen des Aortenstamms, Skoliose und Pectusdeformitäten teilt. Allerdings wird LDS oft durch aggressivere und weitreichendere arterielle Erkrankungen, einzigartige kraniofaziale Merkmale wie Hypertelorismus oder eine gespaltene Uvula sowie das Fehlen von Ektopie lentis (luxierte Linse) unterschieden, was ein Schlüsselmerkmal von MFS ist. Darüber hinaus können bestimmte Typen des Ehlers-Danlos-Syndroms (EDS) ähnliche Befunde aufweisen; beispielsweise kann die kyphoskoliotische EDS eine „marfanoide Habitus“ aufweisen, während die hypermobile EDS mit Aortendilatation assoziiert sein kann. Phänotypen wie MASS (Mitralklappe, Aorta, Haut und Skelett) werden ebenfalls als verwandte, aber mildere Erkrankung im MFS-Spektrum betrachtet.
In welchem Alter bemerkt man das Marfan-Syndrom?
Obwohl Menschen mit Marfan-Syndrom geboren werden, können sich die Anzeichen und Symptome in jedem Alter, von der Kindheit bis ins Erwachsenenleben, bemerkbar machen. Die Merkmale der Erkrankung, wie ein großer, schlanker Körperbau und überproportional lange Gliedmaßen, werden oft während der Wachstumsschübe in der Kindheit und Jugend deutlicher. Infolgedessen variiert das Diagnosealter erheblich; Studien zeigen, dass das mediane Alter für die Diagnose bei etwa 19 Jahren liegt, viele Personen jedoch erst weit ins Erwachsenenalter identifiziert werden. Da die klinischen Anzeichen subtil sein können und im Laufe der Zeit auftreten, wird manchmal gesagt, dass einige Menschen „in die Diagnose hineinwachsen“, wenn sie älter werden und die Merkmale deutlicher werden.
Welche Gesichtsmerkmale hat eine Person mit Marfan-Syndrom?
Personen mit Marfan-Syndrom können mehrere ausgeprägte Gesichtsmerkmale aufweisen, die zu einem erkennbaren Erscheinungsbild beitragen. Ihr Schädel kann lang und schmal sein ( Dolichocephalie ), mit anderen Merkmalen wie tief sitzenden Augen ( Enophthalmos ), flachen Wangenknochen ( Malarhypoplasie ) und einem abwärts gerichteten Augenwinkel. Es ist auch üblich, dass diese Personen einen kleinen Kiefer ( Mikrognathie ) haben, der zurückgesetzt sein kann ( Retrognathie ). Im Mund sind häufig ein stark gewölbter Gaumen und engstehende Zähne zu beobachten, die manchmal zu Problemen bei der Ausrichtung der oberen und unteren Zähne beim Beißen führen können. Während nicht jeder Mensch all diese Merkmale hat und deren Schwere stark variieren kann, ist diese Kombination charakteristisch für die Erkrankung.
Welche Zahnprobleme haben Personen mit Marfan-Syndrom?
Personen mit Marfan-Syndrom haben oft eine Vielzahl von zahnärztlichen und oralen Problemen, die sich aus den Auswirkungen der Erkrankung auf das Bindegewebe ergeben. Ein sehr häufiges Merkmal ist ein hoher, gewölbter Gaumen, der häufig zu schweren Zahnengständen und fehlerhaften Bissen (Malokklusion), wie z.B. einem hinteren Kreuzbiss, führt. Die Zähne selbst können unüblich lang und schmal sein, mit möglichen Wurzelveränderungen oder strukturellen Defekten im Zahnschmelz und Dentin. Innerhalb der Zähne besteht eine höhere Prävalenz von Pulpakalzifikationen oder "Pulpensteinen". Diese strukturellen Herausforderungen, insbesondere das Platzproblem, können eine effektive Mundhygiene erschweren, was zu einer höheren Anfälligkeit für Parodontalerkrankungen wie Gingivitis und Parodontitis beiträgt.
Was ist der Fingertest für das Marfan-Syndrom?
Die Fingertests für das Marfan-Syndrom sind einfache körperliche Manöver, die verwendet werden, um nach überproportional langen Fingern (Arachnodaktylie) zu suchen. Der erste ist das Daumenzeichen (oder Steinberg-Zeichen), bei dem Sie eine Faust machen, während Sie den Daumen innen halten; ein positives Zeichen tritt auf, wenn die Spitze des Daumens über die Kante Ihrer Hand hinausragt. Der zweite ist das Handgelenkzeichen (oder Walker-Murdoch-Zeichen), bei dem Sie Ihren Daumen und kleinen Finger um Ihr anderes Handgelenk wickeln; es gilt als positiv, wenn Ihr Daumen und kleiner Finger sich überlappen. Diese Zeichen deuten auf die lange, schlanke Knochenstruktur hin, die charakteristisch für den Zustand ist. Während sie allein nicht ausschlaggebend sind, sind diese Befunde wichtige Indikatoren, die von Kliniken während einer körperlichen Untersuchung auf Marfan-Syndrom verwendet werden.
Wie unterscheidet man zwischen Ehlers-Danlos und Marfan-Syndrom?
Die Unterscheidung zwischen Marfan-Syndrom und Ehlers-Danlos-Syndrom (EDS) beruht auf ihren jeweiligen primären Merkmalen und den zugrunde liegenden genetischen Ursachen. Marfan-Syndrom, verursacht durch eine Mutation im FBN1 Gen, wird klassisch durch eine Kombination von Skelettproblemen (große Statur, lange Gliedmaßen), kardiovaskulären Problemen (insbesondere Aneurysmen des Aortenstamms) und einer sehr spezifischen Augenkrankheit namens Ektopie lentis (luxierte Linse) definiert, die kein Merkmal von EDS ist. Im Gegensatz dazu handelt es sich bei EDS um eine Gruppe von Erkrankungen, die das Kollagen betreffen, wobei charakteristische Anzeichen eine signifikante Gelenkhypermobilität, Hauthyperextensibilität und fragile Gewebe sind, die zu atrophen Narben führen. Während beide Erkrankungen überlappende Merkmale wie Gelenkhypermobilität und aortale Probleme aufweisen können, zeigt das Vorhandensein von Ektopie lentis stark auf das Marfan-Syndrom hin, während extreme Hautfragilität auf EDS hindeutet. Genetische Tests werden häufig verwendet, um eine Diagnose zu bestätigen und zwischen beiden zu unterscheiden.
