


Die Ursachen des Marfan-Syndroms: Ein genetischer Tiefenblick
Was ist das Marfan-Syndrom?
Das Marfan-Syndrom ist eine genetische Störung, die das Bindegewebe des Körpers betrifft, das starke, flexible Material, das Ihre Organe und Strukturen unterstützt und verbindet. Betrachten Sie das Bindegewebe als den inneren "Kleber und Gerüst" des Körpers, das überall von Ihren Knochen bis zu Ihren Blutgefäßen Stärke und Elastizität bietet. Wenn dieses Material von Geburt an fehlerhaft ist, kann es zu einer Vielzahl von gesundheitlichen Herausforderungen führen, die mehrere Systeme betreffen.
Die sichtbarsten Anzeichen erscheinen oft im Skelett und führen zu einem charakteristisch hohen, schlanken Körperbau mit ungewöhnlich langen Gliedmaßen und hochflexiblen Gelenken. Auch Sehprobleme sind häufig, einschließlich eines signifikanten Risikos, dass sich die Linse des Auges verschiebt. Die schwerwiegendsten Komplikationen betreffen jedoch das Herz-Kreislauf-System. Das geschwächte Bindegewebe kann dazu führen, dass die Aorta – die größte Arterie des Körpers – sich dehnt und sich wölbt, was zu einem potenziell tödlichen Riss führen kann. Das Verständnis der Grundursache dieser weit verbreiteten Schwäche ist der Schlüssel zur Bewältigung der Störung.
Die Hauptursache: Eine Mutation im FBN1-Gen
Im Kern des Marfan-Syndroms steht ein einzelner genetischer Fehler. Die Erkrankung wird durch eine Mutation in einem Gen verursacht, das als FBN1 bekannt ist. Dieses Gen liefert die Blaupause für ein Protein namens Fibrillin-1, einen kritischen Bestandteil des Bindegewebes des Körpers. Wenn das FBN1 Gen mutiert ist, wird die Produktion von Fibrillin-1 gestört, was eine Kaskade von Problemen auslöst, die die strukturelle Integrität des Körpers vom molekularen Niveau an schwächen.
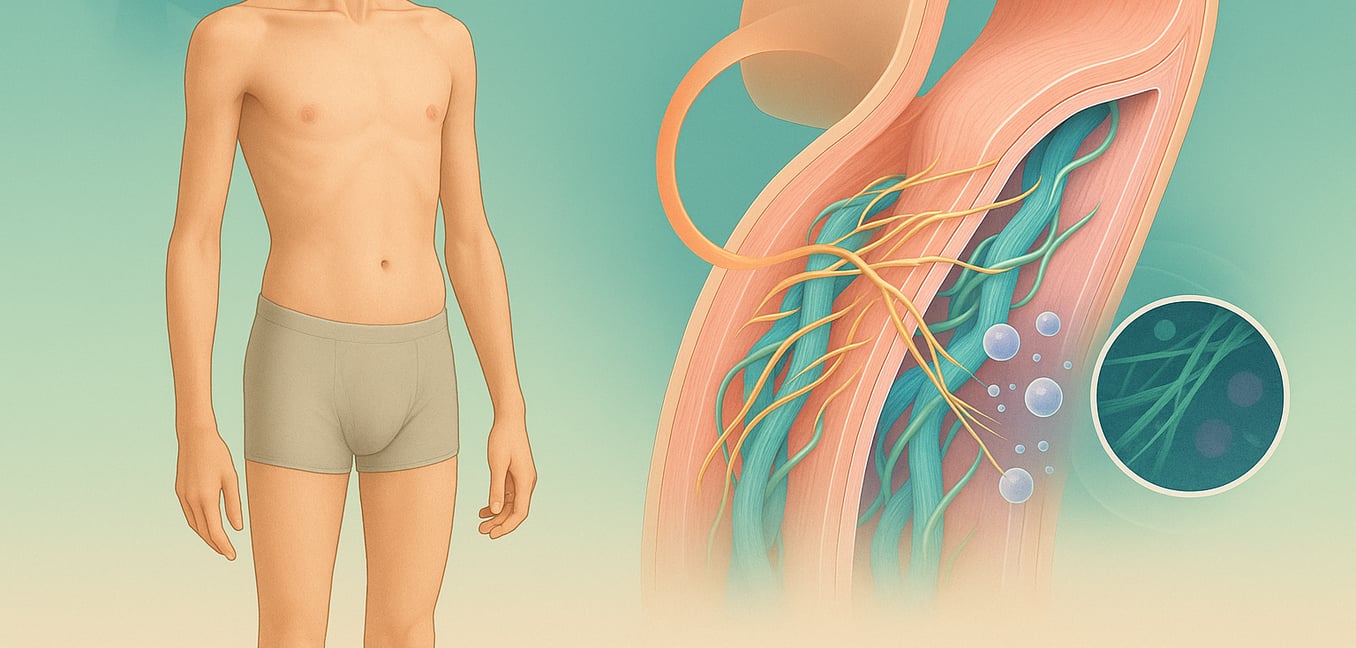
Fibrillin-1: Das fehlerhafte Gerüst des Körpers
Die Hauptaufgabe von Fibrillin-1 besteht darin, mikroskopisch kleine Fäden zu bilden, die Mikrofibrillen genannt werden. Diese Mikrofibrillen fungieren als ein komplexes Gerüstsystem innerhalb der Gewebe und bieten einen Rahmen, auf dem ein anderes Protein, Elastin, abgelagert wird. Zusammen bilden Fibrillin-1 und Elastin die elastischen Fasern, die es den Geweben in der Aorta, der Haut und den Lungen ermöglichen, sich zu dehnen und ohne Schaden zurückzufedern.
Wenn das FBN1 Gen mutiert ist, ist das resultierende Fibrillin-1-Protein defekt. Das, was Genetiker als eine "dominant negative" Mutation bezeichnen. Es bedeutet, dass das fehlerhafte Protein nicht nur versagt, seine Aufgabe zu erfüllen, sondern aktiv das normale Protein stört, das von der gesunden Kopie des Gens produziert wird. Wie eine einzige verdorbene Zutat, die ein ganzes Rezept ruiniert, wird das defekte Fibrillin-1 in die Mikrofibrillenstrukturen eingewebt und beeinträchtigt die Integrität des gesamten Netzwerks. Dies schafft ein grundlegend schwaches und ungeordnetes Gerüst, das direkt zur Gewebeschwäche im ganzen Körper führt.
Das TGF-β-Problem: Ein unkontrolliertes Wachsts-Signal
Über seine strukturelle Rolle hinaus hat das Mikrofibrillennetzwerk eine weitere kritische Funktion: die Regulierung des Wachstums. Diese Strukturen fungieren als Lagerräume, die ein kraftvolles Wachstums-Signal-Molekül namens Transformierendes Wachstumsfaktor Beta (TGF-β) sicher einlagern und inaktiv halten.
Beim Marfan-Syndrom verlieren die defekten Mikrofibrillen ihre Fähigkeit, TGF-β richtig zu halten. Das Schloss auf dem Lagertank ist gebrochen, wodurch übermäßige Mengen dieses potenten Wachstums-Signals in das umgebende Gewebe strömen können. Dies hinterlässt ein kraftvolles "Wachse und remodeliere"-Signal, das in der "ein"-Position feststeckt. Wissenschaftler verstehen mittlerweile, dass dieses überaktive TGF-β-Signal ein wesentlicher Treiber der Marfan-Pathologie ist. Es fördert Entzündungen und löst die Produktion von Enzymen aus, die aktiv Gewebe abbauen, und schafft so einen Teufelskreis, der die aortale Wand weiter schwächt und zu dem charakteristischen Skelettwachstum des Syndroms beiträgt.
Wie die Erkrankung entsteht: Vererbung und spontane Mutation
Eine Person kann das Marfan-Syndrom auf zwei Arten entwickeln: durch Vererbung des fehlerhaften Gens von einem Elternteil oder durch eine neue, spontane genetische Mutation. Das Verständnis beider Wege ist entscheidend für die Familienplanung und genetische Beratung.
Familiäre Vererbung: Das Gen weitergeben
In etwa 75% der Fälle ist das Marfan-Syndrom ein Familienangelegenheit. Die Erkrankung wird durch einen "autosomal dominanten" Vererbungsmuster weitergegeben. "Autosomal" bedeutet, dass das FBN1 Gen sich auf einem nicht geschlechtlichen Chromosom befindet, sodass die Störung Männer und Frauen gleichermaßen betrifft. "Dominant" bedeutet, dass das Erben nur einer Kopie des mutierten Gens von einem einzigen Elternteil ausreicht, um die Erkrankung zu verursachen.
Für einen Elternteil mit Marfan-Syndrom hat jedes Kind, das sie haben, eine 50%ige Chance, das fehlerhafte Gen zu erben, eine Wahrscheinlichkeit, die sich mit jeder Schwangerschaft zurücksetzt. Es ist wichtig zu beachten, dass die Schwere der Erkrankung stark variieren kann, selbst unter Mitgliedern derselben Familie. Ein Elternteil mit relativ milden Merkmalen kann ein Kind mit schwereren Komplikationen haben und umgekehrt.
Spontane Mutation: Wenn es mit Ihnen beginnt
In den verbleibenden 25% der Fälle tritt das Marfan-Syndrom bei einer Person ohne Familiäre Geschichte der Störung auf. Dies wird als spontane oder "de novo" Mutation bezeichnet. Der genetische Fehler im FBN1 Gen tritt zum ersten Mal bei dieser Person auf, normalerweise als zufälliges Ereignis während der Bildung der Spermien- oder Eizelle, die sie erschaffen hat.
Da die Mutation neu ist, sind die Eltern nicht betroffen und tragen das Gen nicht. Aus medizinischer Sicht macht es keinen Unterschied, wo die Mutation ihren Ursprung hat – ob vererbt oder spontan. Die betroffene Person sieht sich denselben Gesundheitsrisiken gegenüber und benötigt dieselbe lebenslange Überwachung und Behandlung. Sobald diese spontane Mutation jedoch aufgetreten ist, trägt diese Person das fehlerhafte Gen jetzt in allen ihren Zellen und kann es mit derselben 50%-Wahrscheinlichkeit an ihre eigenen Kinder weitergeben. So wird die Störung in eine neue Familieneinheit eingeführt.
