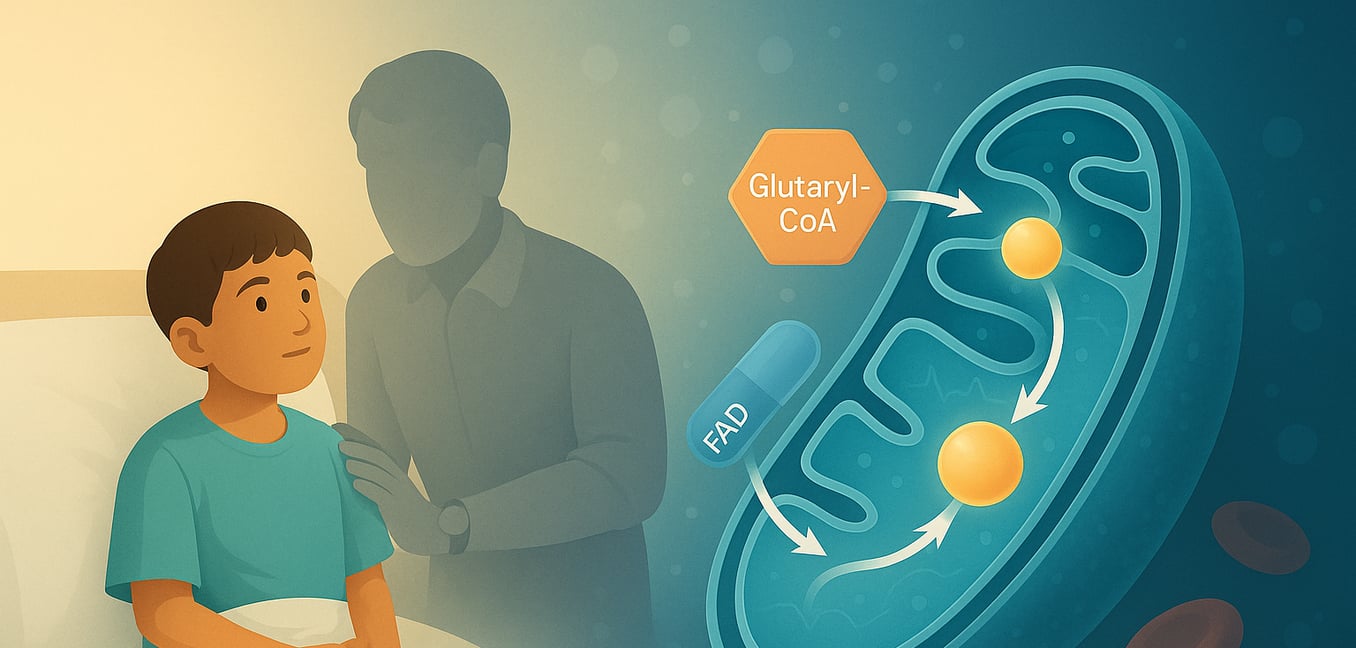



La glutaryl-CoA deshidrogenasa (GCDH) es una enzima vital ubicada dentro de las mitocondrias, las centrales eléctricas de nuestras células. Codificado por el GCDH gen, esta proteína pertenece a la familia de las acil-CoA deshidrogenasas (ACD) y juega un papel crítico en las vías de descomposición metabólica de tres aminoácidos esenciales: L-lisina, L-hidroxilisina y L-triptófano. El correcto funcionamiento de GCDH es crucial para prevenir la acumulación de sustancias potencialmente dañinas en el cuerpo. Cuando esta enzima es deficiente o no funciona correctamente debido a mutaciones genéticas, conduce a un trastorno metabólico grave conocido como aciduria glutarica tipo I (GA1), subrayando la importancia de su actividad para la salud en general, particularmente el desarrollo neurológico.
La función principal de GCDH es catalizar una reacción química específica: la descarboxilación oxidativa de glutaryl-CoA en crotonil-CoA y dióxido de carbono. Este proceso complejo ocurre en varios pasos distintos dentro de la matriz mitocondrial, donde GCDH existe como un homotetrámero (un complejo de cuatro subunidades idénticas). La reacción comienza cuando el sustrato glutaryl-CoA se une a la forma oxidada de la enzima. Un residuo de aminoácido crucial en GCDH, glutamato-370 (Glu370), actúa como una base catalítica, abstrayendo un protón (el protón alfa) del sustrato. Luego, un ion hidruro se transfiere desde el carbono beta del sustrato a la posición N(5) de la dinucleótido de flavina adenina (FAD), un coenzima asociado a GCDH. Esta transferencia reduce FAD a FADH2. Este paso facilita la descarboxilación (remoción de un grupo carboxilo como CO2) de un intermedio unido a la enzima, glutaconil-CoA, rompiendo su enlace Cγ-Cδ. Esta ruptura da lugar a la formación de un anión dienolato, un protón y dióxido de carbono. El intermedio dienolato se protona luego, dando como resultado el producto final, crotonil-CoA, que es posteriormente liberado del sitio activo de la enzima junto con CO2. Para completar el ciclo catalítico y regenerar la enzima para otra ronda, el FADH2 reducido se re-oxidiza de nuevo a FAD transfiriendo sus electrones en dos pasos de electrones individuales a un aceptador de electrones externo, típicamente la flavoproteína que transfiere electrones (ETF).
Comprender la función de GCDH es primordial porque su deterioro tiene consecuencias severas. En individuos con GA1, las mutaciones en el GCDH gen conducen a una deficiencia en la enzima GCDH funcional. Como resultado, el cuerpo no puede procesar eficazmente el glutaryl-CoA. Este bloqueo en la vía metabólica provoca la acumulación de glutaryl-CoA y sus metabolitos avanzados. Estos se convierten en otros compuestos, principalmente ácido glutarico (GA), ácido 3-hidroxiglutarico (3-OH-GA) y, en menor medida, ácido glutacónico, que se acumulan en los fluidos y tejidos corporales. Estos ácidos orgánicos acumulados son neurotóxicos, causando daño particular a regiones específicas del cerebro como los ganglios basales, que son cruciales para controlar el movimiento. Esta neurotoxicidad es responsable de los síntomas característicos de GA1, incluidos crisis encefalopáticas agudas, distonía y otros trastornos severos del movimiento. La acumulación de carnitina glutárica (C5DC), un subproducto no tóxico, también es un marcador característico y se utiliza en pruebas de detección neonatal para detectar el trastorno temprano, permitiendo una intervención oportuna para manejar la condición, como por ejemplo, restringir la lisina en la dieta.
¿Qué es una deficiencia de deshidrogenasa de hidroxiacil-CoA de cadena larga?
La deficiencia de deshidrogenasa de hidroxiacil-CoA de cadena larga (LCHAD) es un trastorno metabólico heredado que impide que el cuerpo descomponga eficientemente ciertos tipos de grasas, conocidas como ácidos grasos de cadena larga, para convertirlos en energía. Esta condición ocurre debido a una deficiencia o mal funcionamiento de la enzima LCHAD, que juega un papel crucial en la vía de beta-oxidación de ácidos grasos mitocondriales. Como resultado, estos ácidos grasos de cadena larga no procesados pueden acumularse en el cuerpo, llevando a problemas de salud potencialmente graves que afectan al hígado, corazón, músculos y cerebro, particularmente durante períodos de ayuno o enfermedad. Los síntomas clave pueden incluir hipoglucemia (bajo nivel de azúcar en sangre), debilidad muscular (miopatía) y problemas cardíacos (cardiomiopatía), destacando la necesidad de un diagnóstico temprano y un manejo cuidadoso, a menudo implicando una dieta especial.
¿Cuál es la función del gen PKLR?
El gen PKLR proporciona las instrucciones esenciales para sintetizar la enzima piruvato quinasa, que juega un papel crítico en la producción de energía celular. Esta enzima es un actor clave en la glucólisis, la vía metabólica que descompone la glucosa (un tipo de azúcar) en piruvato, liberando energía en el proceso. El gen PKLR dirige específicamente la producción de dos formas de piruvato quinasa: la tipo L, predominantemente activa en el hígado, y la tipo R, que funciona en los glóbulos rojos. En ambos tejidos, la piruvato quinasa cataliza el paso final, generador de energía de la glucólisis, facilitando la transferencia de un grupo fosfato desde el fosfoenolpiruvato al ADP para formar ATP, la principal moneda energética de la célula.
