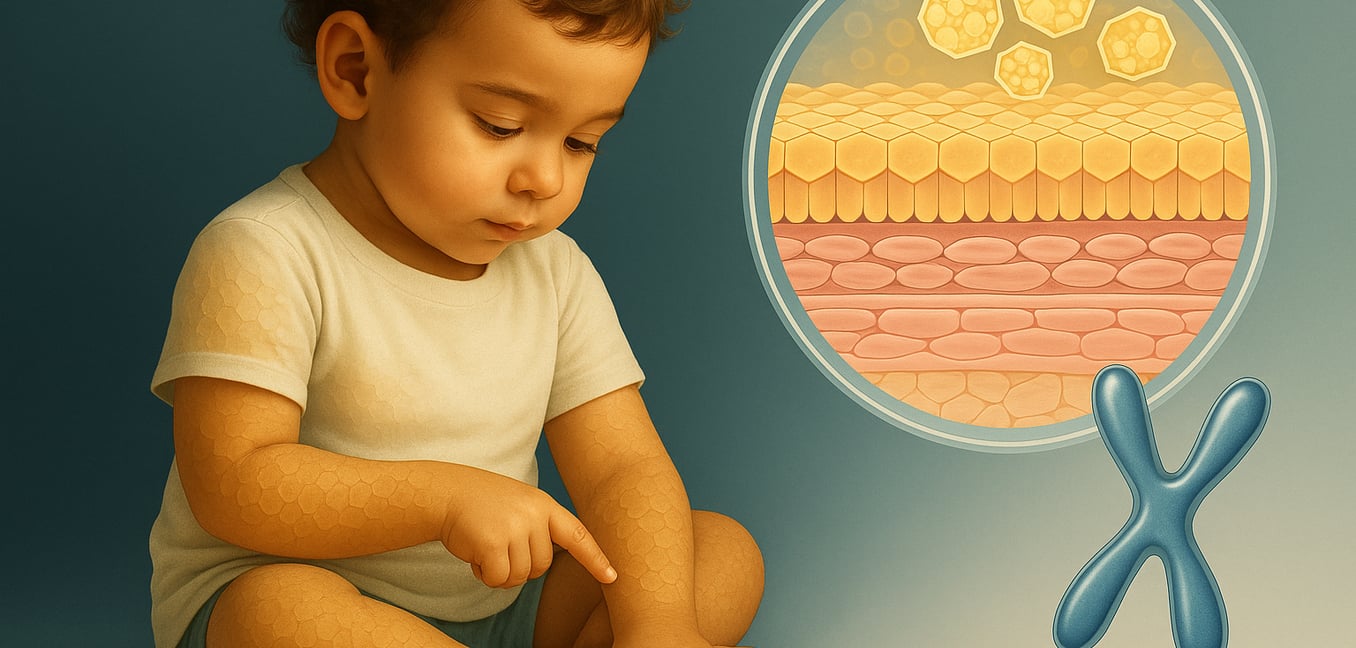



¿Qué es la ictiosis recesiva ligada al cromosoma X?
La ictiosis recesiva ligada al cromosoma X (XLRI) es la segunda forma hereditaria más común de ictiosis, una familia de trastornos de la piel conocidos por causar piel seca y escamosa. La condición afecta casi exclusivamente a los hombres debido a su patrón de herencia ligado al cromosoma X. Los signos típicamente aparecen dentro del primer año de vida, presentándose como escamas marrones en forma de placa que son más prominentes en el torso, el cuello y las extremidades.
Una característica diagnóstica clave es la preservación de los pliegues de la piel, lo que significa que los pliegues de los codos y las rodillas a menudo permanecen claros. Los síntomas tienden a empeorar en los meses fríos y secos del invierno y mejoran con la humedad y la exposición solar del verano. Aunque la condición de la piel es la característica principal, la XLRI también puede asociarse con otros hallazgos de salud, como manchas nubladas inofensivas en la córnea y un mayor riesgo de testículos no descendidos. La condición es causada por una deficiencia en una enzima llamada sulfatasa esteroide, que interrumpe el proceso natural de desprendimiento de la piel.
Las causas genéticas de la XLRI: un gen STS defectuoso
La causa raíz de la ictiosis ligada al cromosoma X es un defecto en el gen de la sulfatasa esteroide (STS), que se encuentra en el brazo corto del cromosoma X. Este gen proporciona las instrucciones para producir la enzima sulfatasa esteroide, una proteína esencial para la salud de la piel. Cuando el gen STS es defectuoso, el cuerpo no puede producir una enzima funcional, lo que lleva directamente a los síntomas de la XLRI. Los errores genéticos responsables caen en dos categorías principales.
Eliminación completa del gen: la causa principal
En la gran mayoría de los casos—hasta el 90%—la XLRI es causada por una eliminación completa del gen STS. En este escenario, el plano genético completo para la enzima falta del cromosoma X. Este tipo de error a gran escala ocurre a menudo durante la formación de células espermáticas o ovocitos. Debido a que la célula no tiene instrucciones que seguir, no puede producir ninguna enzima sulfatasa esteroide, resultando en las características clásicas de la condición. Este cambio estructural grande es típicamente detectable con pruebas genéticas que buscan segmentos faltantes de cromosomas, como un microarray cromosómico.
Mutaciones puntuales: un pequeño error con un gran impacto
En el 10% restante de los individuos, el gen STS está presente pero contiene un pequeño error crítico llamado mutación puntual. Esto es como un solo error tipográfico en el código del gen que hace que las instrucciones sean inutilizables. Estas mutaciones pueden cambiar un componente crucial de la enzima o insertar una señal de "paro" prematura que detiene su producción. Aunque el gen no está completamente faltante, estos sutiles cambios son suficientes para crear una enzima no funcional, lo que lleva al mismo resultado clínico que una eliminación completa. Identificar estos errores más pequeños requiere un análisis genético más detallado, como la secuenciación de ADN.
El impacto bioquímico: cómo la deficiencia enzimática afecta la piel
La enzima sulfatasa esteroide es responsable de descomponer una sustancia cerosa llamada sulfato de colesterol. En la XLRI, la ausencia de esta enzima lleva a una cascada de cambios bioquímicos en la epidermis, la capa más externa de la piel.
Acumulación de sulfato de colesterol
La principal consecuencia de la deficiencia de enzima STS es la acumulación de sulfato de colesterol en el estrato córneo, la capa más externa de la piel. En la piel sana, la enzima mantiene estos niveles bajo control. En la XLRI, las concentraciones pueden volverse de diez a veinte veces más altas de lo normal. Esta acumulación es la causa directa de la descamación de la piel y puede medirse en un análisis de sangre para ayudar a confirmar un diagnóstico.
Fallo en la eliminación natural de la piel
Esta acumulación causa "hiperqueratosis por retención", un término para la incapacidad de la piel de desprender sus células muertas. El proceso natural de desprendimiento, llamado descamación, depende de otras enzimas para disolver el "pegamento" basado en proteínas (corneodesmosomas) que mantiene unidas a las viejas células de la piel. Las investigaciones muestran que el exceso de sulfato de colesterol actúa como un poderoso inhibidor, desactivando esencialmente estas enzimas. Como resultado, el pegamento persiste y las células muertas de la piel permanecen pegadas, formando las gruesas y adherentes escamas características de la XLRI.
Disrupción de la barrera cutánea
La barrera protectora de la piel, que previene la pérdida de agua y bloquea irritantes, depende de una estructura de lípidos altamente organizada. La acumulación anormal de sulfato de colesterol interrumpe este delicado equilibrio, comprometiendo la integridad de la barrera. Esto no solo contribuye a la sequedad crónica de la piel, sino que también altera su función general y permeabilidad.
Casos complejos: síndromes de deleción génica contigua
Mientras que la mayoría de las deleciones genéticas están confinadas al gen STS, algunas son mucho más grandes, eliminando un vecindario completo de genes adyacentes en el cromosoma X. Cuando esto sucede, resulta en un "síndrome de deleción génica contigua", donde un individuo tiene XLRI junto con otras condiciones médicas distintas. Los síntomas específicos dependen completamente de qué genes vecinos también faltan.
Condiciones asociadas en la XLRI sindrómica
La eliminación de genes cerca del STS puede dar lugar a una imagen clínica más compleja. Ejemplos comunes incluyen:
- Estatura baja: Causada por la eliminación del gen SHOX, que es esencial para el desarrollo y crecimiento óseo normal. Esto puede resultar en una condición llamada discondrosteosis de Léri-Weill.
- Síndrome de Kallmann: Resulta de eliminar el gen KAL1, lo que lleva a un sentido del olfato reducido o ausente (anosmia) y pubertad retrasada o ausente.
- Condiciones neurodesarrollacionales: Vinculadas a la eliminación de genes como NLGN4X, aumentando potencialmente el riesgo de discapacidad intelectual, dificultades de aprendizaje o trastorno por déficit de atención e hiperactividad (TDAH).
