



El viaje con una enfermedad rara a menudo comienza con una búsqueda de respuestas. Para individuos y familias que navegan por una Enfermedad de Almacenamiento Lisosomal (EAL), esa búsqueda los lleva al mundo microscópico de nuestras propias células. Sabemos que este paisaje puede ser intimidante de explorar, lleno de términos científicos complejos y una profunda incertidumbre. Nuestro objetivo es caminar este camino contigo, desglosando la ciencia en información clara y de apoyo. Creemos en el poder de la comprensión, y nuestro compromiso es proporcionar un espacio donde puedas encontrar claridad, comunidad y la tranquilidad de que no estás solo en este viaje.
¿Qué es una Enfermedad de Almacenamiento Lisosomal?

En su esencia, una Enfermedad de Almacenamiento Lisosomal (EAL) es un desafío dentro del "centro de reciclaje" vital de la célula, un orgánulo llamado lisosoma. La historia de las EAL comienza con el innovador trabajo del científico belga Christian de Duve, quien descubrió el lisosoma en la década de 1950, un logro que le valió un Premio Nobel en 1974. Identificó estos pequeños orgánulos como el sistema digestivo de la célula, llenos de proteínas especializadas llamadas enzimas. En una célula sana, los lisosomas están llenos de estas enzimas que descomponen materiales complejos, como grasas y azúcares, en componentes más simples que la célula puede reutilizar (Platt, 2018).
Para alguien con una EAL, una mutación genética específica significa que una de estas enzimas cruciales está ausente o no funciona correctamente. La mayoría de las EAL son autosome recesivas, lo que significa que un niño debe heredar una copia del gen mutado de ambos padres para tener la condición (Meikle et al., 1999).
Imagina un sistema de reciclaje de una ciudad donde el equipo responsable de procesar plásticos deja de funcionar de repente. El plástico comenzaría a acumularse, causando problemas en toda la ciudad. Esto es similar a lo que ocurre dentro de las células en una EAL. El material específico (llamado sustrato) que la enzima faltante debía procesar comienza a acumularse (Platt, 2018). Con el tiempo, esta acumulación hace que los lisosomas se llenen de este material almacenado, lo que daña las células. Esta "crisis de basura celular" puede eventualmente llevar a una amplia gama de síntomas que afectan a los órganos de todo el cuerpo (Parenti, Andria, & Ballabio, 2015).
La Odisea Diagnóstica: Un Espectro Diverso de Condiciones

Los investigadores han identificado más de 70 tipos distintos de EAL. Mientras que todos ellos tienen su origen en un problema dentro del lisosoma, se manifiestan de diversas maneras, cada una definida por la enzima específica que falta y el sustrato que, en consecuencia, se acumula. Algunas de las EAL más reconocidas incluyen:
Enfermedad de Gaucher: Causada por una deficiencia de la enzima glucocerebrosidasa, lo que lleva a la acumulación de una sustancia grasa llamada glucocerebrósido (Grabowski & Hopkin, 2022).
Enfermedad de Pompe: Involucra la acumulación de un azúcar complejo, glucógeno, afectando particularmente a las células musculares debido a la falta de la enzima ácido α-glucosidasa (Kishnani et al., 2006).
Enfermedad de Fabry: Resulta de una deficiencia en la enzima α-galactosidasa A, causando que un tipo de grasa llamada globotriaosilceramida se acumule en células de todo el cuerpo (Germain, 2010).
Mucopolisacaridosis (MPS): Un grupo de trastornos relacionados (incluyendo Hurler, Hunter y Síndrome de Sanfilippo) donde faltan diferentes enzimas necesarias para descomponer moléculas de azúcar complejas llamadas glicosaminoglicanos (GAGs) (Parenti, Andria, & Ballabio, 2015).
Enfermedad de Niemann-Pick (Tipos A, B, C) & Enfermedad de Tay-Sachs: Otras EAL bien conocidas que afectan la capacidad del cuerpo para metabolizar grasas (lípidos), conduciendo a daños neurológicos severos (Platt, 2018).
Debido a que los lisosomas están presentes en prácticamente todas las células, los síntomas de las EAL son extraordinariamente diversos y progresivos. Pueden afectar el cerebro y el sistema nervioso, el corazón, el sistema esquelético, el hígado, el bazo, los pulmones e incluso los ojos (Parenti, Andria, & Ballabio, 2015). Entendemos que cada viaje es diferente, y la gravedad y la edad de inicio pueden variar drásticamente, incluso entre individuos con la misma condición nombrada.
Navegando el Camino hacia una Respuesta
Para muchas familias, el camino hacia un diagnóstico es un proceso largo y arduo, una experiencia a menudo llamada una "odisea diagnóstica" (The LSD Collaborative, 2015). Los síntomas tempranos pueden ser sutiles, no específicos y pueden imitar enfermedades infantiles más comunes, lo que lleva a retrasos frustrantes y diagnósticos erróneos (Wraith, 2002). Sabemos lo difícil que puede ser este período de incertidumbre, lo que subraya el profundo desafío de la conciencia entre aquellos que no están especializados en estas condiciones raras. Un estudio reveló que casi el 89% de los médicos de atención primaria encuestados nunca habían considerado una EAL como un posible diagnóstico para alguno de sus pacientes, lo que subraya el profundo desafío de la conciencia.
El alivio de recibir finalmente un diagnóstico, de tener un nombre para los síntomas desconcertantes y dolorosos, puede ser inmenso. Como compartió una paciente, P., que vive con la enfermedad de Fabry, sus síntomas comenzaron en la infancia, pero no fue diagnosticada hasta décadas más tarde, después de que ya hubiera ocurrido un sufrimiento significativo en su familia.
Los métodos diagnósticos se han vuelto cada vez más sofisticados. Normalmente involucran:
Pruebas de enzimas: Pruebas de sangre que miden el nivel de actividad de enzimas lisosomales específicas. Un nivel significativamente reducido o ausente es un indicador clave de una EAL (Sun, 2018).
Pruebas genéticas: Confirman el diagnóstico al identificar la mutación específica en el gen responsable de la enzima, lo que a veces puede ayudar a predecir la gravedad de la enfermedad o guiar las opciones de tratamiento (Parenti, Andria, & Ballabio, 2015).
Cribado de recién nacidos (NBS): En los últimos años, un número creciente de regiones ha añadido EAL tratables como Pompe, Gaucher, Fabry y MPS I a sus paneles de cribado estándar de recién nacidos (Burton, 2017). Al identificar estas condiciones en los primeros días de vida, antes de que se produzcan daños significativos e irreversibles, el NBS ofrece la mejor oportunidad posible para cambiar el curso de la enfermedad.
El Costo Humano: Más que solo una Gráfica Médica
Sabemos que el impacto de una EAL se extiende mucho más allá de los síntomas físicos. Estas son condiciones crónicas, progresivas y a menudo debilitantes que imponen una profunda carga médica, emocional, social y financiera a los pacientes y sus familias. Las realidades diarias pueden incluir la gestión de dolor crónico y fatiga debilitante. Ted, un hombre que vive con la enfermedad de Gaucher, describió una fatiga inmovilizante que era completamente diferente del cansancio ordinario, haciendo que incluso las tareas simples parecieran monumentales.
El paisaje emocional es complejo, lleno del estrés de manejar una enfermedad crónica, la incertidumbre del futuro y el duelo que puede acompañar la salud en declive. Como compartió Jennie, la madre de Sophia, que tiene la enfermedad de Pompe, el tiempo después del diagnóstico fue aterrador: "La parte más aterradora... no tener respuestas definitivas y no saber qué podría pasar o cuándo." Es un viaje que reconfigura las dinámicas familiares y las decisiones de vida, y estamos comprometidos a proporcionar una comunidad donde estas experiencias profundamente personales puedan ser compartidas, validadas y comprendidas.
Una Idea se Convierte en Realidad
Durante décadas, las opciones para tratar estas condiciones fueron desgarradoramente limitadas. Pero una idea revolucionaria, conceptualizada por primera vez en 1964 por el descubridor del lisosoma, Christian de Duve, cambió todo (Beck, 2018). El concepto era simple pero profundo: ¿y si pudiéramos reemplazar la enzima faltante desde el exterior? Esta idea sentó las bases para la Terapia de Reemplazo Enzimático (TRE) (de Duve, 1964).
El viaje desde el concepto hasta la clínica fue uno de enorme dedicación científica. En la década de 1970, experimentos cruciales mostraron que las células deficientes en un plato de laboratorio podrían absorber las enzimas faltantes secretadas por células sanas, un fenómeno llamado "corrección cruzada." (Fratantoni, Hall, & Neufeld, 1968). Esto demostró que el principio básico podía funcionar. Más investigaciones identificaron la "etiqueta de dirección" celular que guía las enzimas hacia el lisosoma: una molécula de azúcar llamada manosa-6-fosfato (M6P) que se une a receptores específicos de M6P en la superficie celular (Kornfeld, 1986).
El verdadero avance clínico fue liderado por el Dr. Roscoe Brady y su equipo en los Institutos Nacionales de Salud (Beck, 2018). Centrándose en la enfermedad de Gaucher, purificaron la enzima necesaria (glucocerebrosidasa) de placentas humanas y la modificaron bioquímicamente para dirigirse a las células correctas (Brady et al., 1974). Los ensayos clínicos fueron un éxito impresionante, llevando a la primera aprobación de la FDA para una TRE, Ceredase™, en 1991 (Beck, 2018). Esto marcó el comienzo de una nueva era.
Hoy, gracias a la tecnología de ADN recombinante, las TRE ya no se derivan de tejido humano. Se producen de manera segura y consistente en grandes cantidades en entornos de laboratorio seguros, un avance importante que ha mejorado la seguridad y el suministro (Platt, 2018).
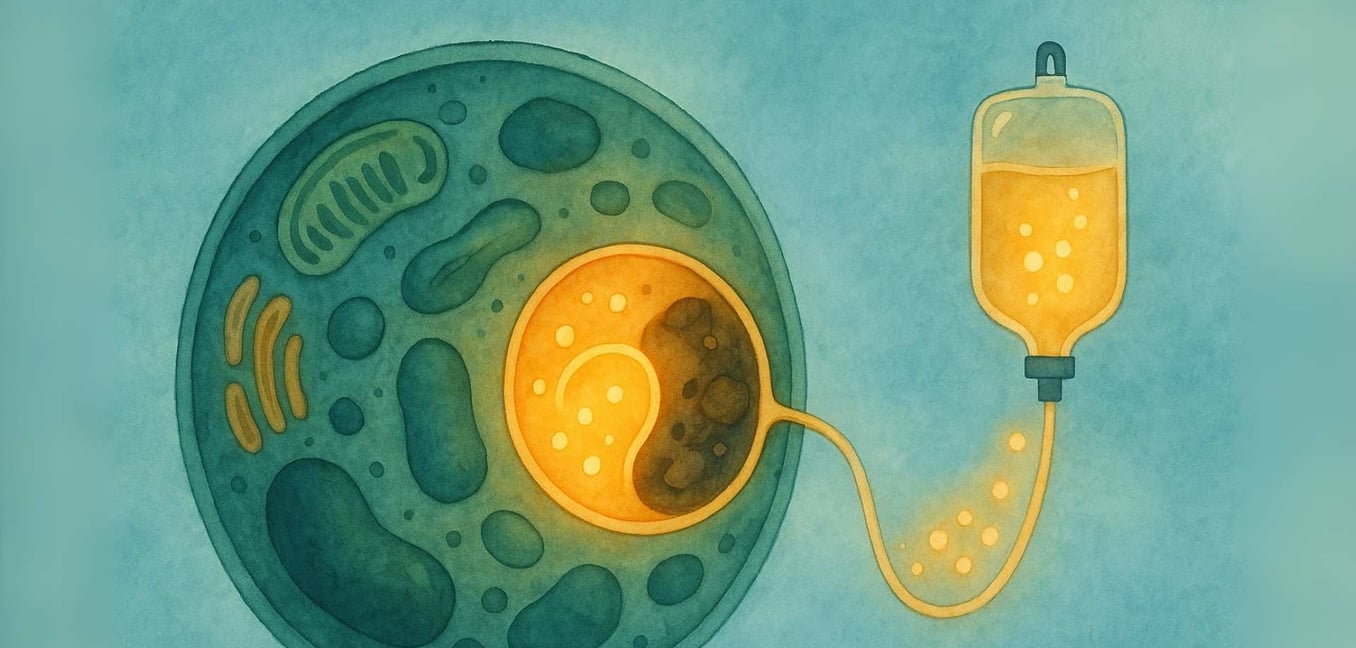
Cómo Funciona la Terapia de Reemplazo Enzimático (TRE)

La TRE es un tratamiento médico diseñado para entregar una versión funcional de la enzima faltante directamente a las células que más la necesitan. Aquí hay un vistazo más cercano al proceso:
La Infusión: La terapia se administra por vía intravenosa (IV), generalmente en una infusión lenta durante varias horas, cada una o dos semanas (Beck, 2018).
El Sistema de Entrega: Estas enzimas se diseñan con esa especial etiqueta "M6P" que las células del cuerpo pueden reconocer. Esto permite que la enzima se una a los receptores de M6P en la superficie celular, actuando como una llave que encaja en una cerradura específica (Kornfeld, 1986).
Acción Celular: Una vez que la célula reconoce y se une a la enzima, la da la bienvenida en su interior a través de un proceso llamado endocitosis y la guía directamente a los lisosomas. Allí, la enzima de reemplazo puede finalmente comenzar su trabajo, descomponiendo los materiales acumulados y ayudando a restaurar un equilibrio más saludable dentro de la célula (Kornfeld, 1986).
El Camino del Tratamiento: Esperanza y Expectativas Realistas
La TRE es un tratamiento, no una cura (Platt, 2018). No corrige el defecto genético subyacente y requiere un compromiso de por vida con infusiones regulares, una parte significativa de la vida para pacientes y sus familias (Beck, 2018).
Además, la TRE enfrenta varios obstáculos significativos:
La Barrera Hematoencefálica (BHE): Este es, sin duda, el mayor desafío. La BHE es un escudo protector que impide la entrada de moléculas grandes, incluyendo las enzimas TRE, en el cerebro. Dado que se estima que dos tercios de las EAL tienen síntomas neurológicos, la TRE estándar a menudo no logra abordar el deterioro cognitivo u otros problemas del SNC, incluso a medida que ayuda al resto del cuerpo.
La Respuesta Inmune: Debido a que la enzima infundida es una proteína, el cuerpo a veces puede verla como "extraña" y desarrollar anticuerpos anti-fármaco (ADAs). Estos anticuerpos pueden, en algunos casos, reducir la efectividad de la terapia o causar reacciones asociadas a la infusión (IARs), como fiebre, sarpullido o respuestas alérgicas más graves.
Entrega a "Sitios Santuario": Más allá del cerebro, ciertos tejidos son difíciles de alcanzar para las grandes moléculas de enzima. Estos incluyen hueso denso, cartílago avascular y válvulas cardíacas, lo que significa que los problemas esqueléticos y las enfermedades valvulares del corazón pueden progresar a pesar del tratamiento.
Expandiendo el Arsenal de Terapias

Las limitaciones de la TRE han inspirado a los investigadores y a nuestra comunidad a presionar incansablemente por nuevas y mejores terapias. El futuro del tratamiento se centra en un ataque múltiple contra estas condiciones, con varias estrategias emocionantes emergiendo:
Terapia de Reducción de Sustrato (TRS): En lugar de reemplazar la enzima, este enfoque utiliza un medicamento oral para inhibir una enzima involucrada en la producción del sustrato en primer lugar. Esto efectivamente "aligera la carga" en los lisosomas comprometidos. Medicamentos aprobados como Miglustat y Eliglustat se utilizan para la enfermedad de Gaucher tipo 1.
Terapia de Chaperona Farmacológica (TCF): Para pacientes cuyos cuerpos producen una enzima que está mal plegada pero aún tiene cierta función potencial, estos pequeños medicamentos orales actúan como un "andamiaje". Se unen a la enzima inestable, ayudándola a plegarse correctamente para que pueda pasar el control de calidad de la célula y llegar al lisosoma para cumplir su función. Migalastat es una chaperona aprobada para los pacientes de Fabry con mutaciones "aptas".
Transplante de Células Madre Hematopoyéticas (TCMH): Para algunas EAL severas con involucramiento del SNC, como MPS I (síndrome de Hurler), el TCMH (o trasplante de médula ósea) de un donante sano puede ser una opción. La idea es que las células del donante viajarán al cerebro y proporcionarán una fuente local y continua de la enzima faltante. Sin embargo, este es un procedimiento de muy alto riesgo con complicaciones potenciales significativas.
Terapia Génica: Este es uno de los frentes más emocionantes y potencialmente transformadores. El objetivo es entregar una copia funcional del gen correcto a las propias células de un paciente, permitiendo que produzcan la enzima por sí mismas. Usando vectores virales diseñados o otros sistemas de entrega, los investigadores están explorando formas de proporcionar un tratamiento duradero y potencialmente único que corrija la causa raíz de la enfermedad. Se están llevando a cabo ensayos clínicos para Pompe, Fabry y varios trastornos de MPS, ofreciendo una poderosa fuente de esperanza.
Avanzando Juntos
El camino desde el descubrimiento fundamental del lisosoma hasta el desarrollo de la TRE y el amanecer de la medicina basada en genes es un testimonio del poder de la investigación científica y la resiliencia humana. Estamos comprometidos a continuar este avance hacia adelante, juntos. Siempre nos esforzaremos por proporcionar información clara y confiable, para fomentar una comunidad de apoyo y cuidado, y para promover la investigación que ofrezca esperanzas tangibles para un futuro más brillante.
Para obtener una visión concisa pero perspicaz de la Terapia de Reemplazo Enzimático, te invitamos a sintonizar nuestro nuevo episodio de podcast. Está diseñado para desglosar este tema complejo en segmentos fácilmente digeribles.
https://youtu.be/T91yim2x8ac?si=Rla7FYMAWxCLUVc8
Referencias
Nobel Prize Outreach AB. (2024). El Premio Nobel en Fisiología o Medicina 1974. NobelPrize.org.
de Duve, C. (2013). Christian de Duve: Explorador de la célula que descubrió nuevos orgánulos utilizando una centrífuga. Proceedings of the National Academy of Sciences, 110(31), 12559-12561.
The Rockefeller University. (n.d.). "Explorando Células con una Centrípeta": El Descubrimiento del Lisosoma. Hospital Centennial.
MSD Manual Profesional. (n.d.). Visión General de los Trastornos de Almacenamiento Lisosomal.
Columbia University Irving Medical Center. (n.d.). Trastornos de Almacenamiento Lisosomal Pediátricos.
Cleveland Clinic. (2022, 27 de junio). Enfermedades y Trastornos de Almacenamiento Lisosomal.
Cleveland Clinic. (2022, 27 de junio). Enfermedades y Trastornos de Almacenamiento Lisosomal.
Bioscience Institute. (n.d.). Enfermedades de Almacenamiento Lisosomal (EAL).
van der Meijden, J. C., et al. (2010). 'Doctor Google' poniendo fin a la odisea diagnóstica en los trastornos de almacenamiento lisosomal. Archives of Disease in Childhood, 95(8), 642-644.
Medicover Genetics. (2025, 26 de febrero). La odisea diagnóstica: La búsqueda de respuestas para enfermedades raras.
Rare Diseases South Africa. (2023, 28 de febrero). Abordando los desafíos del diagnóstico y tratamiento de las enfermedades de almacenamiento lisosomal.
Greenwood Genetic Center. (2024, 14 de agosto). Trastornos de Almacenamiento Lisosomal: Enfermedad de Gaucher y Fabry.
Wikipedia. (n.d.). Terapia de reemplazo enzimático.
Beck, M. (2018). Terapia de reemplazo enzimático y más allá—en memoria de Roscoe O. Brady, M.D. (1923–2016). Journal of Inherited Metabolic Disease, 41(1), 3-13.
Park, J. J., & Lee, K. (2022). Manosa-6-fosfato glicosil para la orientación lisosomal: varias aplicaciones desde la terapia de reemplazo enzimático hasta quimeras dirigidas a lisosomas. Animal Cells and Systems, 26(3), 84-91.
U.S. Food & Drug Administration (FDA). (1991). Búsqueda de Designaciones y Aprobaciones de Medicamentos Huérfanos: Ceredase.
National Gaucher Foundation. (n.d.). El 25º Aniversario de la Aprobación de la TRE por la FDA.
Begley, D. J. (2015). Modificando el transporte a través de la barrera hematoencefálica para brindar esperanza a los pacientes con enfermedades de almacenamiento lisosomal. Journal of Cerebral Blood Flow & Metabolism, 35(1), 3-5.
Pardridge, W. M. (2015). Orientación a la Barrera Hematoencefálica de las Enzimas Lisosomales Terapéuticas. Enfermedades de Almacenamiento Lisosomal, 327-343.
Aflaki, E., et al. (2018). Terapias de reemplazo enzimático: ¿cuál es la mejor opción?. Current Pharmaceutical Design, 24(11), 1238-1250.
Giugliani, R., et al. (2018). Terapia de reemplazo enzimático para mucopolisacaridosis: nuevos desarrollos y resultados clínicos. Expert Opinion on Orphan Drugs, 6(4), 277-287.
Gaucher Disease News. (n.d.). Terapia de reducción de sustrato para la enfermedad de Gaucher.
National Gaucher Foundation. (n.d.). Terapia de Reducción de Sustrato.
Shin, S-H., et al. (2007). Las chaperonas farmacológicas corrigen el almacenamiento lisosomal en la enfermedad de Fabry causada por variantes incompetentes de tráfico. American Journal of Physiology-Cell Physiology, 292(5), C1879-C1887.
Fabry Disease News. (2024, 19 de abril). Terapia de chaperona para la enfermedad de Fabry.
National MPS Society. (n.d.). TCMH.
La-Fauci, G., & A.H. Schuchman. (2016). Terapia Génica para Trastornos de Almacenamiento Lisosomal: Avances Recientes y Limitaciones. Journal of Inborn Errors of Metabolism and Screening, 4, 1-7.
Iimori, T., et al. (2023). Terapia génica para enfermedades de almacenamiento lisosomal: Prospectos actuales de ensayos clínicos. Frontiers in Genetics, 14, 1064924.
Platt, F. M., d'Azzo, A., Davidson, B. L., Neufeld, E. F., & Tifft, C. J. (2018). Enfermedades de almacenamiento lisosomal. Nature Reviews Disease Primers, 4(1), 27.