


Una Introducción al Síndrome de Marfan
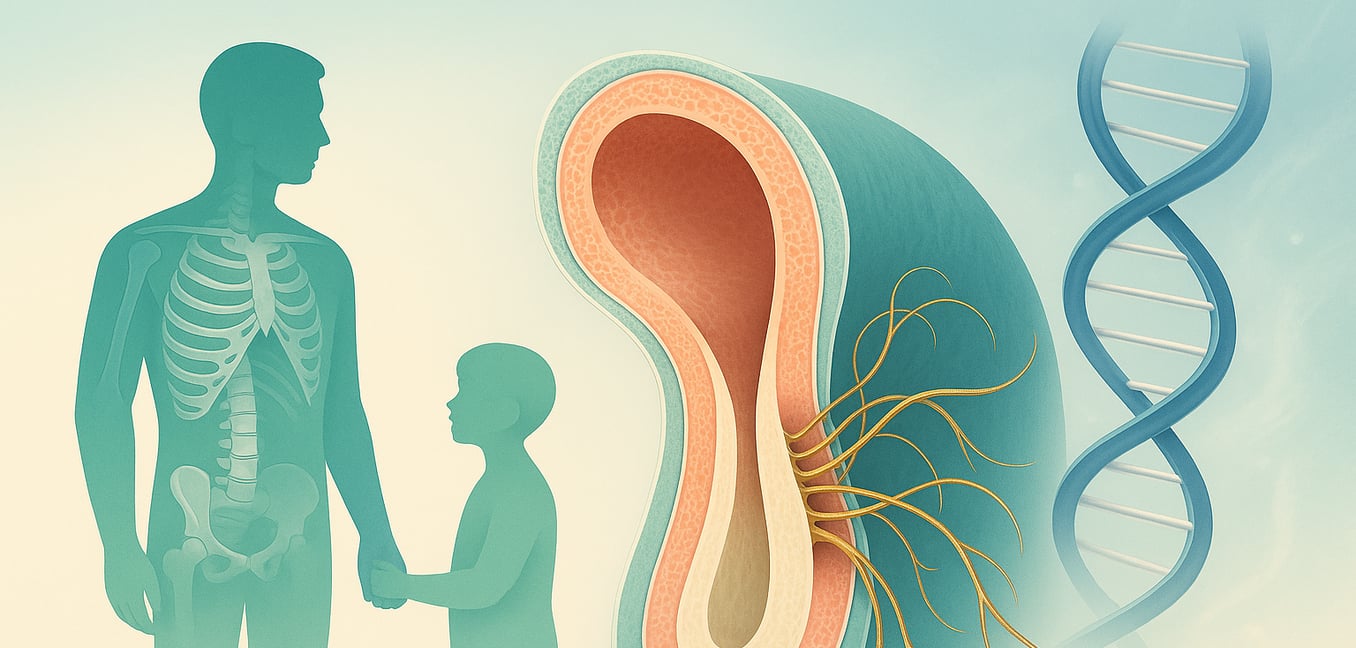
El síndrome de Marfan es un trastorno genético que afecta el tejido conectivo del cuerpo, el material esencial que actúa como un pegamento y andamiaje para órganos, vasos sanguíneos, huesos y músculos. Dado que el tejido conectivo está presente en todo el cuerpo, la condición puede llevar a una amplia y variada gama de desafíos de salud, afectando notablemente el esqueleto, el corazón y los ojos.
Las personas con el síndrome suelen ser inusualmente altas y delgadas, con extremidades y dígitos largos. También pueden desarrollar una columna vertebral curvada, conocida como escoliosis, o un pecho que se hunde hacia adentro o protruye. Sin embargo, las complicaciones más amenazantes para la vida involucran el sistema cardiovascular, particularmente la aorta—la principal arteria del cuerpo. El tejido debilitado puede hacer que la aorta se estire y agrande, creando un alto riesgo de una ruptura repentina y peligrosa. El impacto del síndrome es altamente variable; incluso dentro de la misma familia, algunos individuos pueden tener solo rasgos físicos leves mientras que otros enfrentan complicaciones severas y progresivas, lo que subraya la necesidad de un manejo médico cuidadoso.
El Gen FBN1[^2] : De Plano a Malfuncionamiento
En el corazón del síndrome de Marfan hay una mutación en un solo gen: FBN1. Ubicado en el cromosoma 15, este gen contiene las instrucciones para fabricar fibrilina-1, una proteína grande que es un componente crítico para el tejido conectivo del cuerpo. Las proteínas de fibrilina-1 se ensamblan en estructuras parecidas a hilos llamadas microfibrillas, que forman una malla de apoyo alrededor de las células. Estas microfibrillas confieren a tejidos como la aorta su elasticidad y proporcionan soporte estructural a partes más rígidas del cuerpo, como los huesos y los ligamentos que sostienen el lente del ojo en su lugar. Un gen FBN1 defectuoso interrumpe todo este proceso, llevando a la debilidad sistémica observada en el síndrome de Marfan.
Cómo las Mutaciones Interrumpen la Fibrilina-1
Un cambio en el gen FBN1 puede comprometer el tejido conectivo de dos maneras principales:
Producir muy poca proteína (Haploinsuficiencia). Algunas mutaciones resultan en que el cuerpo produzca una cantidad insuficiente de fibrilina-1 funcional. En este escenario, uno de los dos copias del gen FBN1 no funciona, por lo que el cuerpo solo puede depender de la única copia saludable. Esto no es suficiente para satisfacer las necesidades del cuerpo. Piense en ello como tratar de construir una pared de ladrillos con solo la mitad de los ladrillos requeridos; la estructura resultante es inherentemente débil e incompleta.
Producir una proteína defectuosa e interferente (Efecto Dominante-Negativo). Otras mutaciones hacen que el gen produzca una proteína de fibrilina-1 alterada y malformada que sabotea activamente la proteína normal. Esta proteína defectuosa se incorpora a la estructura de la microfibrilla junto a la proteína saludable. Esto es como mezclar ladrillos en mal estado con los buenos; los componentes defectuosos comprometen la integridad de toda la pared, a menudo llevando a problemas estructurales más severos.
Los Efectos Posteriores en el Cuerpo
La descomposición en la formación de microfibrillas tiene consecuencias que se extienden más allá de la simple debilidad estructural. Estas fibras de soporte tienen una función secundaria crucial: actúan como unidades de almacenamiento para proteínas que regulan el crecimiento, siendo el Factor de Crecimiento Transformante-beta (TGF-β) el más notable. Normalmente, las microfibrillas mantienen el TGF-β secuestrado e inactivo hasta que se necesita para procesos como la reparación y desarrollo de tejidos.
Cuando las mutaciones en FBN1 hacen que las microfibrillas se desorganizen y fragmenten, ya no pueden retener efectivamente el TGF-β. Esto lleva a una cantidad excesiva de TGF-β activo siendo liberado en los tejidos. Esta actividad excesiva se cree que es un motor clave de algunas de las características más reconocibles del síndrome de Marfan, incluida el crecimiento excesivo de los huesos largos que resulta en una constitución alta y delgada.
Patrones de Herencia: Cómo se Transmite el Síndrome de Marfan
El síndrome de Marfan sigue un patrón de herencia autosómica dominante, lo que explica por qué puede afectar a varias generaciones de una familia, pero también aparecer inesperadamente en un individuo sin antecedentes familiares. Comprender este patrón es clave para el asesoramiento genético y la planificación familiar.
- Un gen defectuoso es suficiente. El término "autosomal dominante" significa que el gen FBN1 está en un cromosoma no sexual (autosomal) y solo se necesita una copia mutada de un padre para causar el trastorno (dominante). Afecta a hombres y mujeres por igual.
- Un 50% de probabilidad en cada embarazo. Un padre con síndrome de Marfan tiene un 50/50 de probabilidad de pasar el gen mutado a cada uno de sus hijos. Esta probabilidad es independiente para cada niño, como un lanzamiento de moneda.
- Las nuevas mutaciones son comunes. En aproximadamente el 25% de los casos, la condición surge de una nueva mutación, o "de novo", que ocurre espontáneamente en el óvulo o en la célula del esperma. En estos casos, los padres no están afectados, pero el niño tendrá el síndrome de Marfan y puede pasarlo a sus propios hijos.
- La gravedad no se hereda. Heredar la mutación FBN1 no predice cuán severos serán los síntomas. Un padre con características leves puede tener un hijo con complicaciones mayores, y viceversa. Esta variabilidad hace que el monitoreo médico individualizado sea esencial para todos con la condición.
El Papel de las Pruebas Genéticas en el Diagnóstico
Dado que las características del síndrome de Marfan pueden variar drásticamente y superponerse con otros trastornos del tejido conectivo, las pruebas genéticas se han convertido en una herramienta esencial para proporcionar diagnósticos claros y precisos. Permite a los médicos observar directamente la causa genética subyacente en lugar de depender solo de los síntomas físicos.
- Confirmando el diagnóstico. Cuando los signos clínicos son ambiguos, encontrar una mutación causante de enfermedad conocida en el gen FBN1 puede proporcionar un diagnóstico definitivo, poniendo fin a la incertidumbre para los pacientes y permitiendo a los médicos crear un plan de manejo proactivo.
- Distinguiendo de trastornos similares. Condiciones como el síndrome de Loeys-Dietz pueden imitar el síndrome de Marfan pero son causadas por diferentes mutaciones genéticas y a menudo requieren un tratamiento más agresivo, particularmente para la aorta. Los paneles genéticos que prueban FBN1 y otros genes relacionados aseguran que se haga el diagnóstico correcto.
- Habilitando pruebas predictivas para miembros de la familia. Una vez que se identifica una mutación específica en una familia, otros parientes en riesgo pueden ser evaluados para esa modificación exacta. Esto puede identificar a las personas que tienen la condición antes de que aparezcan los síntomas, permitiendo un monitoreo y tratamiento temprano y que salvan vidas. También libera a los familiares que den resultados negativos de un seguimiento innecesario durante toda la vida.
