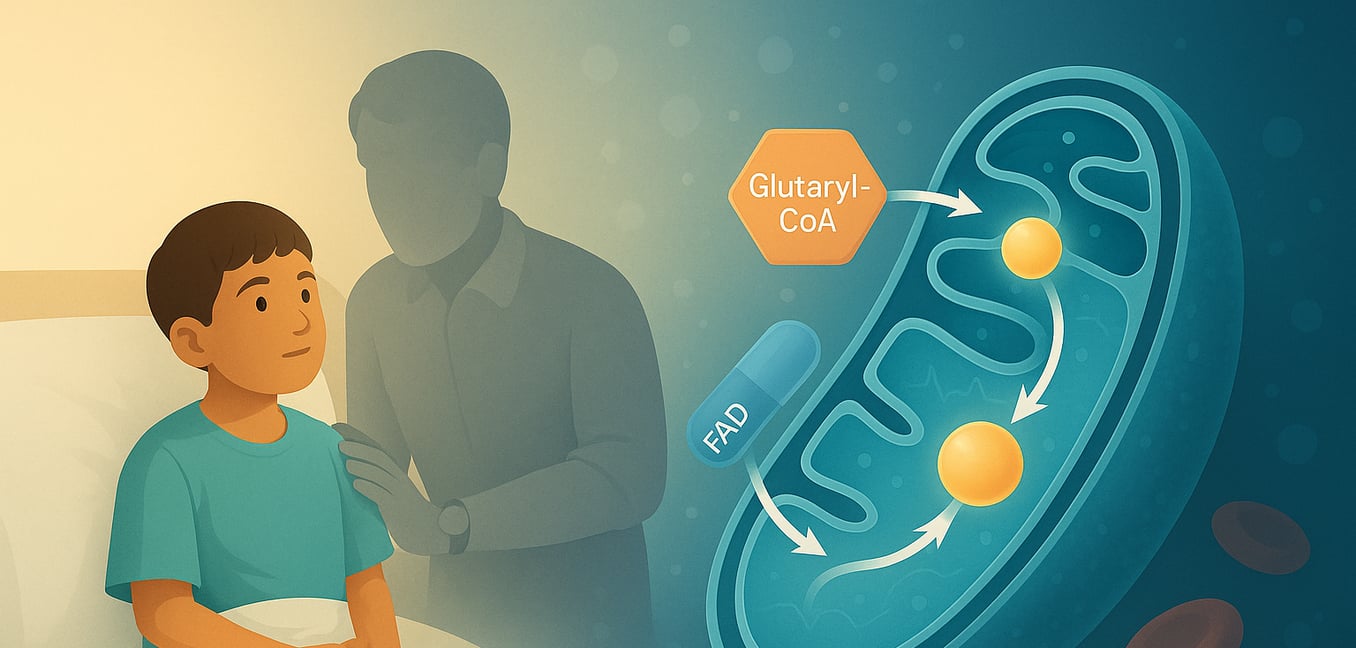



La glutaryle-CoA déshydrogénase (GCDH) est une enzyme vitale située dans les mitochondries, les centrales énergétiques de nos cellules. Codée par le GCDH gène, cette protéine appartient à la famille des déshydrogénases acyles-CoA (ACD) et joue un rôle crucial dans les voies de dégradation métabolique de trois acides aminés essentiels : L-lysine, L-hydroxylysine et L-tryptophane. Le bon fonctionnement de la GCDH est essentiel pour prévenir l'accumulation de substances potentiellement nocives dans le corps. Lorsque cette enzyme est déficiente ou dysfonctionnelle en raison de mutations génétiques, cela entraîne un trouble métabolique grave connu sous le nom d'acidurie glutarique de type I (GA1), soulignant l'importance de son activité pour la santé globale, en particulier le développement neurologique.
La fonction principale de la GCDH est de catalyser une réaction chimique spécifique : la décarboxylation oxydative de la glutaryl-CoA en crotonyl-CoA et dioxyde de carbone. Ce processus complexe se déroule en plusieurs étapes distinctes dans la matrice mitochondriale, où la GCDH existe sous forme d'homotétramère (un complexe de quatre sous-unités identiques). La réaction commence lorsque le substrat glutaryl-CoA se lie à la forme oxydée de l'enzyme. Un résidu d'acide aminé crucial dans la GCDH, le glutamate-370 (Glu370), agit comme une base catalytique, abstrait un proton (le proton alpha) du substrat. Par la suite, un ion hydrure est transféré du carbone bêta du substrat à la position N(5) de la flavine adénine dinucléotide (FAD), un coenzyme lié à la GCDH. Ce transfert réduit le FAD en FADH2. Cette étape facilite la décarboxylation (élimination d'un groupe carboxyle sous forme de CO2) d'un intermédiaire lié à l'enzyme, la glutaconyl-CoA, en rompant son lien Cγ-Cδ. Cette rupture entraîne la formation d'un anion dienolate, d'un proton et de dioxyde de carbone. L'intermédiaire dienolate est ensuite protoné, produisant le produit final, le crotonyl-CoA, qui est ensuite libéré du site actif de l'enzyme avec du CO2. Pour compléter le cycle catalytique et régénérer l'enzyme pour un autre cycle, le FADH2 réduit est ré-oxydé en FAD en transférant ses électrons par étapes de transfert d'électrons uniques à un accepteur d'électrons externe, généralement la flavoprotéine de transfert d'électrons (ETF).
Comprendre la fonction de la GCDH est primordial car son impairment a de graves conséquences. Chez les individus atteints de GA1, des mutations dans le GCDH gène entraînent une carence en enzyme GCDH fonctionnelle. En conséquence, le corps ne peut pas traiter efficacement la glutaryl-CoA. Ce blocage de la voie métabolique entraîne une accumulation de glutaryl-CoA et de ses métabolites en amont. Ceux-ci sont ensuite convertis en d'autres composés, principalement de l'acide glutarique (GA), de l'acide 3-hydroxyglutarique (3-OH-GA) et dans une moindre mesure, de l'acide glutaconique, qui s'accumulent dans les fluides corporels et les tissus. Ces acides organiques accumulés sont neurotoxiques, en particulier dommageables pour des régions spécifiques du cerveau comme les ganglions de la base, qui sont cruciaux pour le contrôle des mouvements. Cette neurotoxicité est responsable des symptômes caractéristiques de la GA1, y compris des crises encéphalopathiques aiguës, de la dystonie et d'autres troubles moteurs graves. L'accumulation de glutarylcarnitine (C5DC), un sous-produit non toxique, est également un indicateur et est utilisé dans les tests de dépistage néonatal pour détecter le trouble tôt, permettant une intervention rapide pour gérer la condition en restreignant par exemple l'apport alimentaire en lysine.
Qu'est-ce qu'une carence en déshydrogénase d'hydroxyacyl-CoA à chaînes longues ?
La carence en déshydrogénase d'hydroxyacyl-CoA à chaînes longues (LCHAD) est un trouble métabolique héréditaire qui empêche le corps de décomposer efficacement certains types de graisses, appelées acides gras à chaînes longues, pour les convertir en énergie. Cette condition survient en raison d'une déficience ou d'une dysfonction de l'enzyme LCHAD, qui joue un rôle crucial dans la voie d'oxydation beta des acides gras mitochondriaux. En conséquence, ces acides gras à chaînes longues non traités peuvent s'accumuler dans le corps, entraînant des problèmes de santé potentiellement graves affectant le foie, le cœur, les muscles et le cerveau, en particulier pendant les périodes de jeûne ou de maladie. Les symptômes clés peuvent inclure une hypoglycémie, une faiblesse musculaire (myopathie) et des problèmes cardiaques (cardiomyopathie), soulignant la nécessité d'un diagnostic précoce et d'une gestion soigneuse, souvent en impliquant un régime alimentaire spécial.
Quelle est la fonction du gène PKLR ?
Le gène PKLR fournit les instructions essentielles pour la synthèse de l'enzyme pyruvate kinase, qui joue un rôle critique dans la production d'énergie cellulaire. Cette enzyme est un acteur clé de la glycolyse, la voie métabolique qui décompose le glucose (un type de sucre) en pyruvate, libérant de l'énergie dans le processus. Le gène PKLR dirige spécifiquement la production de deux formes de pyruvate kinase : le type L, principalement actif dans le foie, et le type R, qui fonctionne dans les globules rouges. Dans les deux tissus, la pyruvate kinase catalyse l'étape finale, génératrice d'énergie de la glycolyse, facilitant le transfert d'un groupe phosphate du phosphoénolpyruvate à l'ADP pour former de l'ATP, la principale monnaie énergétique de la cellule.
