



Le parcours avec une maladie rare commence souvent par une recherche de réponses. Pour les individus et les familles naviguant dans une Maladie de Stockage Lysosomal (MSL), cette recherche vous entraîne dans le monde microscopique de nos propres cellules. Nous savons que cela peut être un paysage intimidant à explorer, rempli de termes scientifiques complexes et d'une profonde incertitude. Notre objectif est de marcher sur ce chemin avec vous, en décomposant la science en informations claires et soutenantes. Nous croyons au pouvoir de la compréhension, et notre engagement est de fournir un espace où vous pouvez trouver clarté, communauté et la reassurance que vous n'êtes pas seul dans ce parcours.
Qu'est-ce qu'une Maladie de Stockage Lysosomal ?

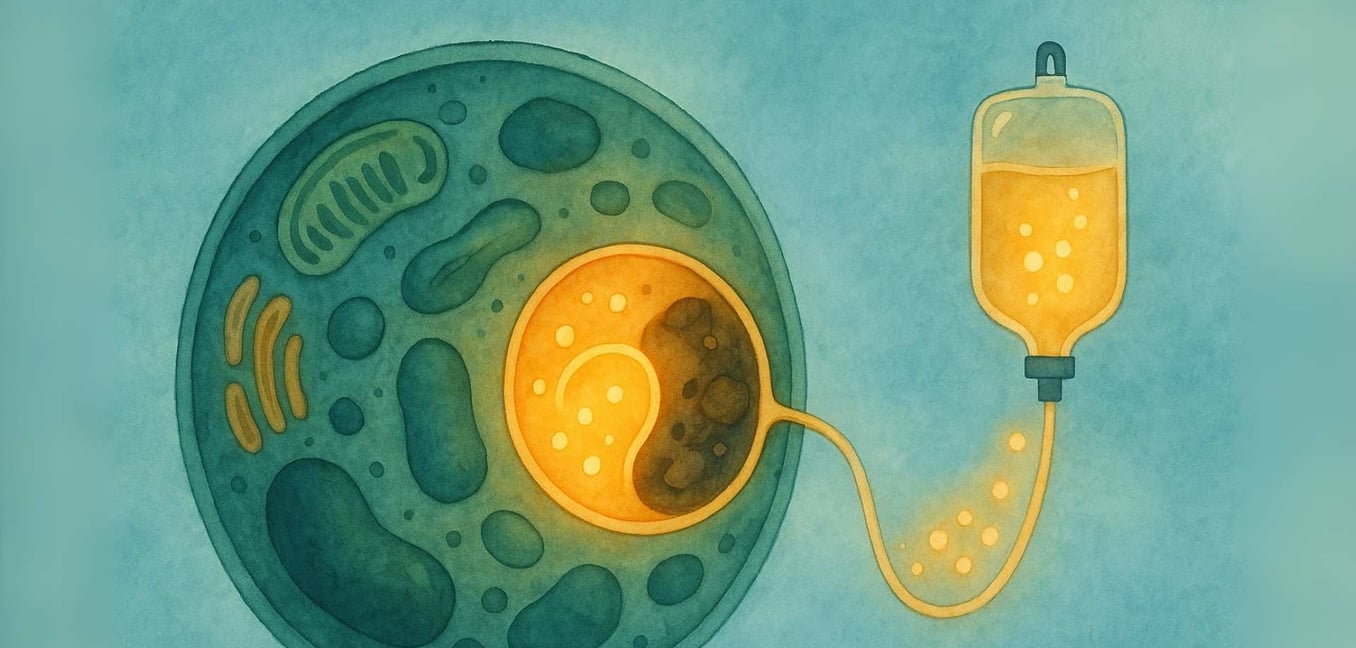
Au cœur, une Maladie de Stockage Lysosomal (MSL) est un défi à l'intérieur du « centre de recyclage » vital de la cellule, un organite appelé lysosome. L'histoire des MSL commence avec le travail révolutionnaire du scientifique belge Christian de Duve, qui a découvert le lysosome dans les années 1950—une réalisation qui lui a valu un Prix Nobel en 1974. Il a identifié ces petits organites comme étant le système digestif de la cellule, remplis de protéines spécialisées appelées enzymes. Dans une cellule saine, les lysosomes sont remplis de ces enzymes qui décomposent des matériaux complexes, comme les graisses et les sucres, en composants plus simples que la cellule peut réutiliser (Platt, 2018).
Pour une personne atteinte d'une MSL, une mutation génétique spécifique signifie que l'une de ces enzymes cruciales est soit absente, soit ne fonctionne pas correctement. La plupart des MSL sont récessives autosomiques, ce qui signifie qu'un enfant doit hériter d'une copie du gène muté des deux parents pour avoir la condition (Meikle et al., 1999).
Imaginez le système de recyclage d'une ville où l'équipe chargée de traiter le plastique cesse soudainement de fonctionner. Le plastique commencerait à s'accumuler, provoquant des problèmes dans toute la ville. C'est similaire à ce qui se passe à l'intérieur des cellules d'une MSL. Le matériau spécifique (appelé substrat) que l'enzyme manquante devait traiter commence à s'accumuler (Platt, 2018). Avec le temps, cette accumulation cause l'engorgement des lysosomes avec ce matériel stocké, ce qui endommage les cellules. Cette "crise des déchets cellulaires" peut finalement conduire à la vaste gamme de symptômes qui affectent les organes tout au long du corps (Parenti, Andria, & Ballabio, 2015).
L'Odyssée Diagnostique : Un Spectre Diversifié de Conditions

Les chercheurs ont maintenant identifié plus de 70 types distincts de MSL. Bien qu'ils proviennent tous d'un problème au sein du lysosome, ils se manifestent de manière très variée, chacun étant défini par l'enzyme spécifique qui manque et le substrat qui s'accumule en conséquence. Parmi les MSL les plus reconnues, on trouve :
Maladie de Gaucher : Causée par une carence en l'enzyme glucocérébrosidase, menant à l'accumulation d'une substance grasse appelée glucocérébroside (Grabowski & Hopkin, 2022).
Maladie de Pompe : Implique l'accumulation d'un sucre complexe, le glycogène, impactant particulièrement les cellules musculaires en raison d'un manque de l'enzyme acide α-glucosidase (Kishnani et al., 2006).
Maladie de Fabry : Résulte d'une carence en l'enzyme α-galactosidase A, provoquant l'accumulation d'un type de graisse appelée globotriaosylcéramide dans les cellules du corps entier (Germain, 2010).
Mucopolysaccharidoses (MPS) : Un groupe de troubles apparentés (y compris les syndromes de Hurler, Hunter, et Sanfilippo) où différentes enzymes nécessaires pour décomposer des molécules de sucre complexes appelées glycosaminoglycanes (GAG) sont manquantes (Parenti, Andria, & Ballabio, 2015).
Maladie de Niemann-Pick (Types A, B, C) & Maladie de Tay-Sachs : D'autres MSL bien connues qui affectent la capacité du corps à métaboliser les graisses (lipides), conduisant à des dommages neurologiques sévères (Platt, 2018).
Parce que les lysosomes sont présents dans presque toutes les cellules, les symptômes des MSL sont extraordinairement divers et progressifs. Ils peuvent impacter le cerveau et le système nerveux, le cœur, le système squelettique, le foie, la rate, les poumons, et même les yeux (Parenti, Andria, & Ballabio, 2015). Nous comprenons que chaque parcours est différent, et la gravité et l'âge d'apparition peuvent varier considérablement, même parmi les individus présentant la même condition nommée.
Naviguer sur le Chemin d'une Réponse
Pour de nombreuses familles, le chemin vers un diagnostic est un processus long et ardu, une expérience souvent décrite comme une "odyssée diagnostique" (The LSD Collaborative, 2015). Les symptômes précoces peuvent être subtils, non spécifiques, et peuvent imiter des maladies infantiles plus courantes, entraînant des retards frustrants et des diagnostics erronés (Wraith, 2002). Nous savons combien cette période d'incertitude peut être difficile, ce qui souligne l'énorme défi de la sensibilisation parmi ceux qui ne sont pas spécialisés dans ces conditions rares. Une étude a révélé que près de 89 % des médecins généralistes interrogés n'avaient même jamais envisagé une MSL comme diagnostic potentiel pour l'un de leurs patients, ce qui souligne l'énorme défi de sensibilisation.
Le soulagement d'obtenir enfin un diagnostic—d'avoir un nom pour les symptômes douloureux et déroutants—peut être immense. Comme l'a partagé une patiente, P., qui vit avec la maladie de Fabry, ses symptômes ont commencé dans son enfance, mais elle n'a pas été diagnostiquée avant des décennies plus tard, après que beaucoup de souffrances aient déjà eu lieu dans sa famille.
Les méthodes diagnostiques sont devenues de plus en plus sophistiquées. Elles impliquent généralement :
Tests d'Enzymes : Tests sanguins qui mesurent le niveau d'activité de certaines enzymes lysosomales. Un niveau significativement réduit ou absent est un indicateur clé d'une MSL (Sun, 2018).
Tests Génétiques : Confirment le diagnostic en identifiant la mutation spécifique dans le gène responsable de l'enzyme, ce qui peut parfois aider à prédire la gravité de la maladie ou à guider les choix de traitement (Parenti, Andria, & Ballabio, 2015).
Dépistage Nouveau-Né (NBS) : Ces dernières années, un nombre croissant de régions ont ajouté des MSL traitables comme Pompe, Gaucher, Fabry et MPS I à leurs panneaux de dépistage néonatal standard (Burton, 2017). En identifiant ces conditions dans les premiers jours de la vie, avant que des dommages significatifs et irréversibles ne puissent se produire, le NBS offre la meilleure opportunité possible de changer le cours de la maladie.
Le Coût Humain : Plus Qu'un Simple Dossier Médical
Nous savons que l'impact d'une MSL va bien au-delà des symptômes physiques. Ce sont des conditions chroniques, progressives et souvent débilitantes qui imposent un lourd fardeau médical, émotionnel, social et financier aux patients et à leurs familles. Les réalités quotidiennes peuvent inclure la gestion de douleurs chroniques et de fatigues débilitantes. Ted, un homme vivant avec la maladie de Gaucher, a décrit une fatigue immobilisante qui était complètement différente de la fatigue ordinaire, rendant même les tâches simples monumentales.
Le paysage émotionnel est complexe, rempli du stress de la gestion d'une maladie chronique, de l'incertitude de l'avenir et du chagrin qui peut accompagner la dégradation de la santé. Comme l'a partagé Jennie, la mère de Sophia qui a la maladie de Pompe, le temps après le diagnostic était terrifiant : "La partie la plus effrayante... était de ne pas avoir de réponses définitives et de ne pas savoir ce qui pouvait arriver ou quand." C'est un parcours qui redessine la dynamique familiale et les choix de vie, et nous nous engageons à fournir une communauté où ces expériences profondément personnelles peuvent être partagées, validées et comprises.
Une Idée Devenant Réalité
Pendant des décennies, les options pour traiter ces conditions étaient désespérément limitées. Mais une idée révolutionnaire, d'abord conceptualisée en 1964 par le découvreur du lysosome, Christian de Duve, a tout changé (Beck, 2018). Le concept était simple mais profond : que se passerait-il si nous pouvions remplacer l'enzyme manquante de l'extérieur ? Cet aperçu a jeté les bases de la Thérapie de Remplacement Enzymatique (TRE) (de Duve, 1964).
Le chemin allant du concept à la clinique était marqué par un immense dévouement scientifique. Dans les années 1970, des expériences cruciales ont montré que des cellules déficientes dans une boîte de pétri pouvaient absorber des enzymes manquantes secrétées par des cellules saines, un phénomène appelé "correction transversale." (Fratantoni, Hall, & Neufeld, 1968). Cela a prouvé que le principe de base pouvait fonctionner. D'autres recherches ont identifié l'"étiquette d'adresse" cellulaire qui guide les enzymes vers le lysosome : une molécule de sucre appelée mannose-6-phosphate (M6P) qui se lie à des récepteurs M6P spécifiques à la surface cellulaire (Kornfeld, 1986).
La véritable percée clinique a été dirigée par Dr. Roscoe Brady et son équipe aux Instituts Nationaux de la Santé (Beck, 2018). Se concentrant sur la maladie de Gaucher, ils ont purifié l'enzyme nécessaire (glucocérébrosidase) à partir de placentas humains et l'ont modifiée biochimiquement pour cibler les bonnes cellules (Brady et al., 1974). Les essais cliniques ont été un succès éclatant, conduisant à la première approbation par la FDA pour une TRE, Ceredase™, en 1991 (Beck, 2018). Cela a marqué le début d'une nouvelle ère.
Aujourd'hui, grâce à la technologie de l'ADN recombinant, les TRE ne sont plus dérivées de tissus humains. Elles sont produites en toute sécurité et de manière constante en grandes quantités dans des laboratoires sécurisés, une avancée majeure qui a amélioré la sécurité et l'approvisionnement (Platt, 2018).
Comment Fonctionne la Thérapie de Remplacement Enzymatique (TRE)

La TRE est un traitement médical conçu pour livrer une version fonctionnelle de l'enzyme manquante directement aux cellules qui en ont le plus besoin. Voici un aperçu du processus :
L'Infusion : La thérapie est administrée par voie intraveineuse (IV), généralement par infusion lente sur plusieurs heures, toutes les une à deux semaines (Beck, 2018).
Le Système de Livraison : Ces enzymes sont conçues avec cette étiquette d'adresse spéciale M6P que les cellules du corps peuvent reconnaître. Cela permet à l'enzyme de se lier aux récepteurs M6P à la surface de la cellule, agissant comme une clé s'insérant dans une serrure spécifique (Kornfeld, 1986).
Action Cellulaire : Une fois que la cellule reconnaît et se lie à l'enzyme, elle l'accueille à l'intérieur par un processus appelé endocytose et la guide directement vers les lysosomes. Là, l'enzyme de remplacement peut enfin entrer en action, décomposant les matériaux accumulés et aidant à restaurer un équilibre plus sain au sein de la cellule (Kornfeld, 1986).
Le Parcours de Traitement : Espoir et Attentes Honnêtes
La TRE est un traitement, pas un remède (Platt, 2018). Elle ne corrige pas le défaut génétique sous-jacent et nécessite un engagement à vie à des infusions régulières, une partie significative de la vie pour les patients et leurs familles (Beck, 2018).
De plus, la TRE fait face à plusieurs défis significatifs :
La Barrière Hémato-encéphalique (BHE) : C'est sans doute le plus grand défi. La BHE est un bouclier protecteur qui empêche les grosses molécules, y compris les enzymes TRE, d'entrer dans le cerveau. Étant donné qu'environ deux tiers des MSL ont des symptômes neurologiques, la TRE standard échoue souvent à traiter le déclin cognitif ou d'autres problèmes du SNC, même alors qu'elle aide le reste du corps.
La Réponse Immune : Comme l'enzyme infusée est une protéine, le corps peut parfois la considérer comme "étrangère" et développer des anticorps anti-médicaments (ADAs). Ces anticorps peuvent, dans certains cas, réduire l'effet du traitement ou causer des réactions associées à l'infusion (IAR), telles que fièvre, éruption cutanée ou réactions allergiques plus sévères.
Livraison aux "Sites Sanctuaires" : Au-delà du cerveau, certains tissus sont difficiles à atteindre pour les grosses molécules enzymatiques. Ceux-ci incluent l'os dense, le cartilage avasculaire et les valves cardiaques, signifiant que des problèmes squelettiques et des maladies valvulaires cardiaques peuvent progresser malgré le traitement.
Élargir l'Arsenal des Thérapies

Les limitations de la TRE ont inspiré les chercheurs et notre communauté à pousser sans relâche pour de nouvelles et meilleures thérapies. L'avenir du traitement est axé sur une attaque à plusieurs volets de ces conditions, avec plusieurs stratégies passionnantes émergentes :
Thérapie de Réduction du Substrat (TRS) : Au lieu de remplacer l'enzyme, cette approche utilise un médicament oral pour inhiber une enzyme impliquée dans la production du substrat en premier lieu. Cela "allège efficacement la charge" sur les lysosomes compromis. Des médicaments approuvés comme Miglustat et Eliglustat sont utilisés pour la maladie de Gaucher de type 1.
Thérapie de Chaperon Pharmacologique (TCP) : Pour les patients dont les corps produisent une enzyme mal pliée mais ayant encore une certaine fonction, ces petits médicaments oraux agissent comme un "échafaudage." Ils se lient à l'enzyme instable, l'aidant à se plier correctement afin qu'elle puisse passer le contrôle de qualité de la cellule et atteindre le lysosome pour faire son travail. Migalastat est un chaperon approuvé pour les patients Fabry avec des mutations "favorables" spécifiques.
Transplantation de Cellules Souches Hématopoïétiques (TCSH) : Pour certaines MSL graves avec atteinte du SNC, comme le MPS I (syndrome de Hurler), la TCSH (ou transplantation de moelle osseuse) à partir d'un donneur en bonne santé peut être une option. L'idée est que les cellules dérivées du donneur se déplacent vers le cerveau et fournissent une source locale et continue de l'enzyme manquante. Toutefois, c'est une procédure à très haut risque avec des complications potentielles significatives.
Thérapie Génique : C'est l'une des frontières les plus passionnantes et potentiellement transformantes. L'objectif est de délivrer une copie fonctionnelle du gène correct dans les propres cellules d'un patient, leur permettant de produire eux-mêmes l'enzyme. En utilisant des vecteurs viraux génétiquement modifiés ou d'autres systèmes de livraison, les chercheurs explorent des moyens de fournir un traitement durable, potentiellement unique, qui corrige la cause racine de la maladie. Des essais cliniques sont en cours pour Pompe, Fabry et plusieurs troubles MPS, offrant une source puissante d'espoir.
Avancer Ensemble
Le parcours allant de la découverte fondamentale du lysosome au développement de la TRE et à l'aube de la médecine basée sur les gènes témoigne du pouvoir de l'enquête scientifique et de la résilience humaine. Nous sommes déterminés à continuer cette avancée collective. Nous nous efforcerons toujours de fournir des informations fiables et claires, de favoriser une communauté solidaire et bienveillante, et de défendre la recherche qui apporte un espoir tangible pour un avenir meilleur.
Pour un aperçu concis mais perspicace de la Thérapie de Remplacement Enzymatique, nous vous invitons à écouter notre nouvel épisode de podcast. Il est conçu pour décomposer ce sujet complexe en segments facilement digestes.
<iframe width="560" height="315" src="https://www.youtube.com/embed/T91yim2x8ac?si=5aRF203Rw9fqjAoN" title="Lecteur vidéo YouTube" frameborder="0" allow="accelerometer; autoplay; clipboard-write; encrypted-media; gyroscope; picture-in-picture; web-share" referrerpolicy="strict-origin-when-cross-origin" allowfullscreen></iframe>
Références
Nobel Prize Outreach AB. (2024). Le Prix Nobel en Physiologie ou Médecine 1974. NobelPrize.org.
de Duve, C. (2013). Christian de Duve : exploreur de la cellule ayant découvert de nouveaux organites en utilisant une centrifugeuse. Proceedings of the National Academy of Sciences, 110(31), 12559-12561.
The Rockefeller University. (n.d.). "Explorer les cellules avec une centrifugeuse" : La découverte du lysosome. Hospital Centennial.
MSD Manual Professional Edition. (n.d.). Aperçu des Troubles de Stockage Lysosomal.
Columbia University Irving Medical Center. (n.d.). Troubles de Stockage Lysosomal Pédiatriques.
Cleveland Clinic. (2022, 27 juin). Maladies et Troubles de Stockage Lysosomal.
Cleveland Clinic. (2022, 27 juin). Maladies et Troubles de Stockage Lysosomal.
Bioscience Institute. (n.d.). Maladies de Stockage Lysosomal (MSLS).
van der Meijden, J. C., et al. (2010). 'Docteur Google' mettant fin à l'odyssée diagnostique dans les maladies de stockage lysosomal. Archives of Disease in Childhood, 95(8), 642-644.
Medicover Genetics. (2025, 26 février). L'odyssée diagnostique : La recherche de réponses pour les maladies rares.
Rare Diseases South Africa. (2023, 28 février). S'attaquer aux défis du diagnostic et du traitement des maladies de stockage lysosomal.
Greenwood Genetic Center. (2024, 14 août). Troubles de Stockage Lysosomal : Maladies de Gaucher et de Fabry.
Wikipedia. (n.d.). Thérapie de remplacement enzymatique.
Beck, M. (2018). Thérapie de remplacement enzymatique et au-delà—en mémoire de Roscoe O. Brady, M.D. (1923–2016). Journal of Inherited Metabolic Disease, 41(1), 3-13.
Park, J. J., & Lee, K. (2022). Glycan mannose-6-phosphate pour le ciblage lysosomal : diverses applications de la thérapie de remplacement enzymatique aux chimères ciblant les lysosomes. Animal Cells and Systems, 26(3), 84-91.
U.S. Food & Drug Administration (FDA). (1991). Recherche de désignations et d'approbations de médicaments orphelins : Ceredase.
National Gaucher Foundation. (n.d.). Le 25e anniversaire de l'approbation par la FDA de la TRE.
Begley, D. J. (2015). Modification du transport à travers la barrière hémato-encéphalique pour apporter de l'espoir aux patients atteints de maladies de stockage lysosomal. Journal of Cerebral Blood Flow & Metabolism, 35(1), 3-5.
Pardridge, W. M. (2015). Ciblage de la barrière hémato-encéphalique des enzymes lysosomales thérapeutiques. Lysosomal Storage Disorders, 327-343.
Aflaki, E., et al. (2018). Thérapies de remplacement enzymatique : quelle est la meilleure option ? Current Pharmaceutical Design, 24(11), 1238-1250.
Giugliani, R., et al. (2018). Thérapie de remplacement enzymatique pour les mucopolysaccharidoses : nouveaux développements et résultats cliniques. Expert Opinion on Orphan Drugs, 6(4), 277-287.
Gaucher Disease News. (n.d.). Thérapie de réduction du substrat pour la maladie de Gaucher.
National Gaucher Foundation. (n.d.). Thérapie de réduction du substrat.
Shin, S-H., et al. (2007). Le chaperon pharmacologique corrige le stockage lysosomal dans la maladie de Fabry causée par des variants incapables de trafic. American Journal of Physiology-Cell Physiology, 292(5), C1879-C1887.
Fabry Disease News. (2024, 19 avril). Thérapie de chaperon pour la maladie de Fabry.
National MPS Society. (n.d.). TCSH.
La-Fauci, G., & A.H. Schuchman. (2016). Thérapie génique pour les troubles de stockage lysosomal : avancées récentes et limitations. Journal of Inborn Errors of Metabolism and Screening, 4, 1-7.
Iimori, T., et al. (2023). Thérapie génique pour les maladies de stockage lysosomal : perspectives des essais cliniques actuels. Frontiers in Genetics, 14, 1064924.
Platt, F. M., d'Azzo, A., Davidson, B. L., Neufeld, E. F., & Tifft, C. J. (2018). Maladies de stockage lysosomal. Nature Reviews Disease Primers, 4(1), 27.