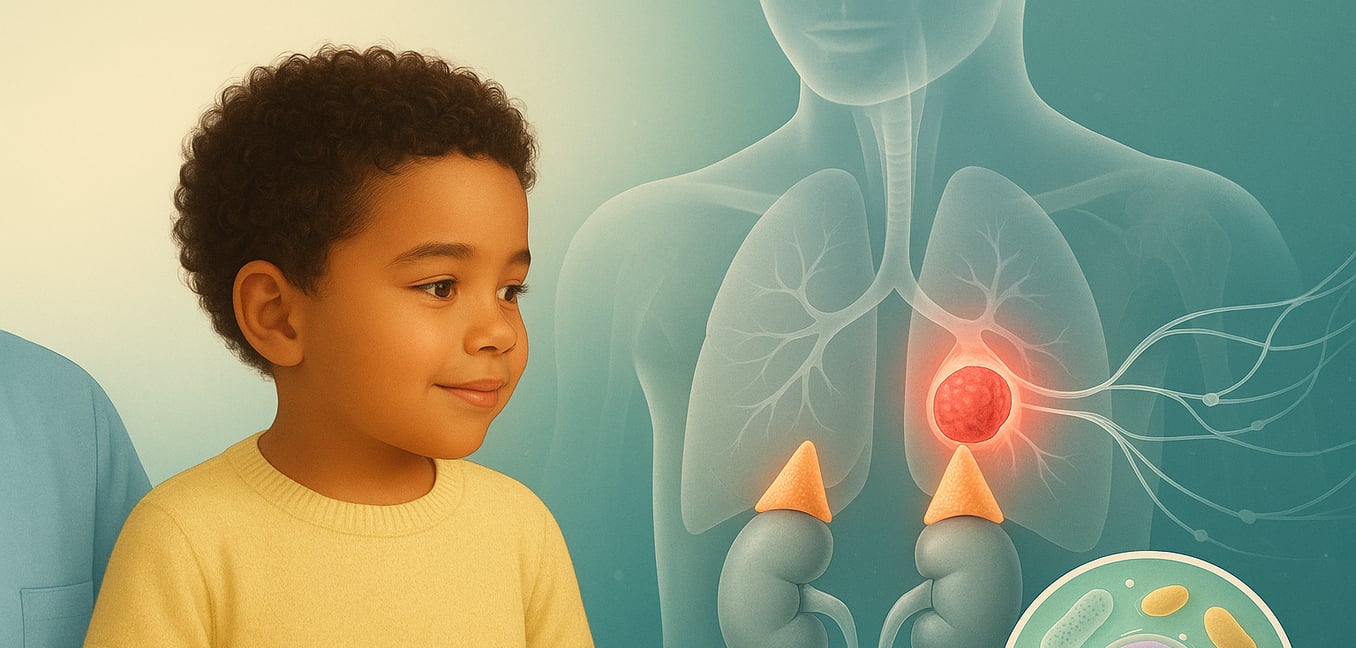



Qu'est-ce que le neuroblastome ?
Le neuroblastome est un cancer des tumeurs solides qui provient de cellules nerveuses immatures appelées neuroblastes. Ce sont les mêmes cellules qui forment le système nerveux sympathique chez un fœtus en développement, le réseau qui contrôle les fonctions automatiques comme la fréquence cardiaque et la pression artérielle. Le nom reflète son origine : "neuro" pour les nerfs et "blastome" pour un cancer des cellules en développement.
Principalement cancer des nourrissons et des jeunes enfants, le neuroblastome peut se former n'importe où le long de la colonne vertébrale où ce tissu nerveux existe. Le site le plus commun se trouve dans les glandes surrénales, qui sont situées au-dessus des reins. Son comportement est notoirement imprévisible. Chez certains nourrissons, une tumeur peut disparaître d'elle-même ou évoluer en tissu bénin sans aucun traitement. Chez les enfants plus âgés, cependant, la maladie est souvent agressive, se développant et se propageant rapidement vers d'autres parties du corps.
Dans plus de 98 % des cas, le neuroblastome est sporadique, ce qui signifie qu'il résulte d'une erreur aléatoire durant le développement fœtal et n'est pas héréditaire. Il n'est pas causé par quelque chose que les parents ont fait ou n'ont pas fait, ni lié à des facteurs environnementaux connus.
Reconnaître les signes et les symptômes
Étant donné que le neuroblastome peut se développer à de nombreux endroits différents, ses symptômes sont variés et peuvent souvent imiter des maladies infantiles courantes, rendant le diagnostic difficile. Les signes qu'un enfant présente dépendent de la taille de la tumeur, de son emplacement, et de savoir si le cancer s'est propagé.
Tuméfactions et gonflements abdominaux
Une masse dure et indolore dans l'abdomen est l'un des signes les plus fréquents, car de nombreuses tumeurs commencent dans les glandes surrénales. Cela peut provoquer un ventre gonflé, une mauvaise appétit ou des plaintes de sensation de satiété. La pression d'une tumeur peut également entraîner de la constipation ou une perte de poids inexpliquée, facilement confondue avec des problèmes digestifs typiques.
Symptômes de la maladie généralisée
Lorsque le neuroblastome se propage (métastase), il voyage souvent vers les os et la moelle osseuse. Cela peut causer des douleurs osseuses, en particulier la nuit, ou une claudication sans blessure évidente. Les enfants peuvent également devenir extrêmement irritable, fatigué, ou pâle à cause de l'anémie (un faible taux de globules rouges). Un signe distinctif du neuroblastome généralisé est l'apparition de cercles sombres, semblables à des ecchymoses, autour des yeux, souvent appelés "yeux de raton laveur", causés par la propagation du cancer vers les os près des orbites oculaires.
Signes spécifiques à la localisation
L'emplacement de la tumeur peut produire des symptômes uniques. Une tumeur dans la poitrine peut comprimer la trachée, provoquant des sifflements ou des difficultés respiratoires. Une masse visible dans le cou peut parfois interférer avec les nerfs, entraînant un paupière tombante ou une pupille plus petite dans un œil. Dans de rares cas, le neuroblastome peut déclencher une condition neurologique appelée syndrome d'opsoclonus-myoclonus-ataxie (OMAS), caractérisé par des mouvements oculaires rapides et incontrôlés, des spasmes musculaires saccadés et une mauvaise coordination.
Le processus de diagnostic
Si le neuroblastome est suspecté, les médecins réaliseront une série de tests pour confirmer le diagnostic, déterminer les caractéristiques de la tumeur, et voir si elle s'est propagée. Cette évaluation étape par étape est essentielle pour planifier le traitement approprié.
L'évaluation diagnostique comprend généralement :
- Tests d'imagerie : Les médecins utilisent des échographies, des tomodensitogrammes (TDM) et des IRM (imagerie par résonance magnétique) pour obtenir des images détaillées de la tumeur. Ces images montrent sa taille et son emplacement exacts et révèlent si elle comprime des organes vitaux ou des vaisseaux sanguins.
- Tests urinaires et sanguins : Ces tests vérifient les niveaux élevés de substances chimiques appelées catécholamines (spécifiquement HVA et VMA) qui sont libérées par les cellules de neuroblastome et excrétées dans l'urine. Des tests sanguins sont également effectués pour vérifier l'anémie et évaluer la fonction globale des organes.
- Biopsie : Un chirurgien retire un petit morceau de tissu tumoral, qui est ensuite examiné au microscope par un pathologiste pour confirmer qu'il s'agit de neuroblastome. Une aspiration et une biopsie de la moelle osseuse (prenant un échantillon de l'os de la hanche) est également une procédure standard pour vérifier si le cancer s'est propagé là-bas.
- Scintigraphie MIBG : Ce test d'imagerie spécialisé est unique au neuroblastome. Une substance appelée MIBG, qui est absorbée par la plupart des cellules de neuroblastome, est attachée à une petite quantité d'iode radioactif et injectée dans le corps. Une caméra spéciale scanne ensuite le corps pour créer une carte montrant la tumeur primaire et d'autres zones où le cancer s'est propagé.
Comprendre le stade et les groupes de risque
Une fois le diagnostic confirmé, les médecins déterminent le stade du cancer et assignent un groupe de risque. Le stade décrit où le cancer est situé dans le corps, tandis que le regroupement par risque prédit son comportement probable. Ensemble, ces classifications sont les facteurs les plus importants pour décider du type et de l'intensité du traitement.
Déterminer le stade
Les médecins déterminent le stade en utilisant des scans d'imagerie pour voir si la tumeur s'est propagée ou touche des parties vitales du corps comme les vaisseaux sanguins majeurs ou la moelle épinière. Une tumeur localisée qui ne s'est pas propagée et qui peut être retirée en toute sécurité est considérée comme un stade inférieur. Si des cellules cancéreuses sont trouvées dans des parties éloignées du corps, telles que les os, le foie, ou les ganglions lymphatiques distants, la maladie est classée comme métastatique (Stade M), qui est le stade le plus avancé.
Analyser la biologie de la tumeur
L'échantillon de biopsie fournit des indices cruciaux sur la "personnalité" de la tumeur. Les pathologistes étudient la génétique de la tumeur et l'apparence des cellules au microscope (histologie). Un des marqueurs biologiques les plus critiques est le gène MYCN. S'il y a trop de copies de ce gène (un état appelé amplification), il agit comme une pédale d'accélérateur pour la croissance du cancer, indiquant une maladie plus agressive. D'autres modifications chromosomiques sont également analysées pour déterminer si la tumeur a une biologie "favorable" ou "défavorable".
Attribution d'un groupe de risque
Enfin, toutes les informations - l'âge de l'enfant, le stade du cancer, et ses caractéristiques biologiques - sont combinées pour placer l'enfant dans un groupe de risque faible, intermédiaire ou élevé. Cette classification guide directement le plan de traitement. Les patients à faible risque n'ont besoin que d'une chirurgie ou même simplement d'une observation attentive. Les patients à risque intermédiaire reçoivent généralement une chimiothérapie et une chirurgie. Le neuroblastome à haut risque nécessite une approche de traitement beaucoup plus intensive et en plusieurs étapes.
Un aperçu des options de traitement
Le traitement du neuroblastome est adapté au groupe de risque spécifique de chaque enfant. L'objectif est de guérir le cancer tout en minimisant les effets secondaires à long terme en adaptant l'intensité de la thérapie à l'agressivité de la maladie.
Chirurgie
Pour les tumeurs localisées, une opération visant à retirer la masse cancéreuse peut être le seul traitement nécessaire. Le chirurgien s'efforce de retirer autant de la tumeur que possible sans nuire aux organes voisins. Dans les cas à haut risque, une chimiothérapie est souvent administrée d'abord pour réduire la tumeur, la rendant plus sûre et plus facile à retirer complètement.
Chimiothérapie
La chimiothérapie utilise des médicaments puissants qui circulent dans le sang pour tuer les cellules cancéreuses dans tout le corps. C'est une pierre angulaire du traitement pour le neuroblastome à risque intermédiaire et élevé, ainsi que pour les cancers qui se sont déjà propagés. Les médicaments sont administrés en cycles, avec des périodes de traitement suivies de périodes de repos pour permettre au corps de récupérer.
Radiothérapie
Ce traitement utilise des faisceaux d'énergie haute, comme des rayons X, pour détruire les cellules cancéreuses. Pour le neuroblastome à haut risque, la radiothérapie est souvent utilisée sur le site de la tumeur d'origine après la chirurgie pour éviter qu'elle ne repousse. Elle peut également être utilisée pour traiter des zones douloureuses où le cancer s'est propagé aux os. Une forme spécialisée, la thérapie MIBG, délivre des radiations directement aux cellules de neuroblastome dans tout le corps.
Thérapies avancées pour la maladie à haut risque
Traiter le neuroblastome à haut risque est un processus long et intensif. Cela implique souvent une chimiothérapie à haute dose suivie d'une transplantation de cellules souches. Ce processus détruit toutes les cellules cancéreuses restantes mais anéantit également la moelle osseuse, qui est ensuite remplacée par les propres cellules souches saines de l'enfant collectées plus tôt. Après la transplantation, les patients reçoivent une immunothérapie, un traitement qui active le système immunitaire du corps pour repérer et détruire toutes les cellules cancéreuses persistantes, ce qui aide à réduire le risque de récidive du cancer.
