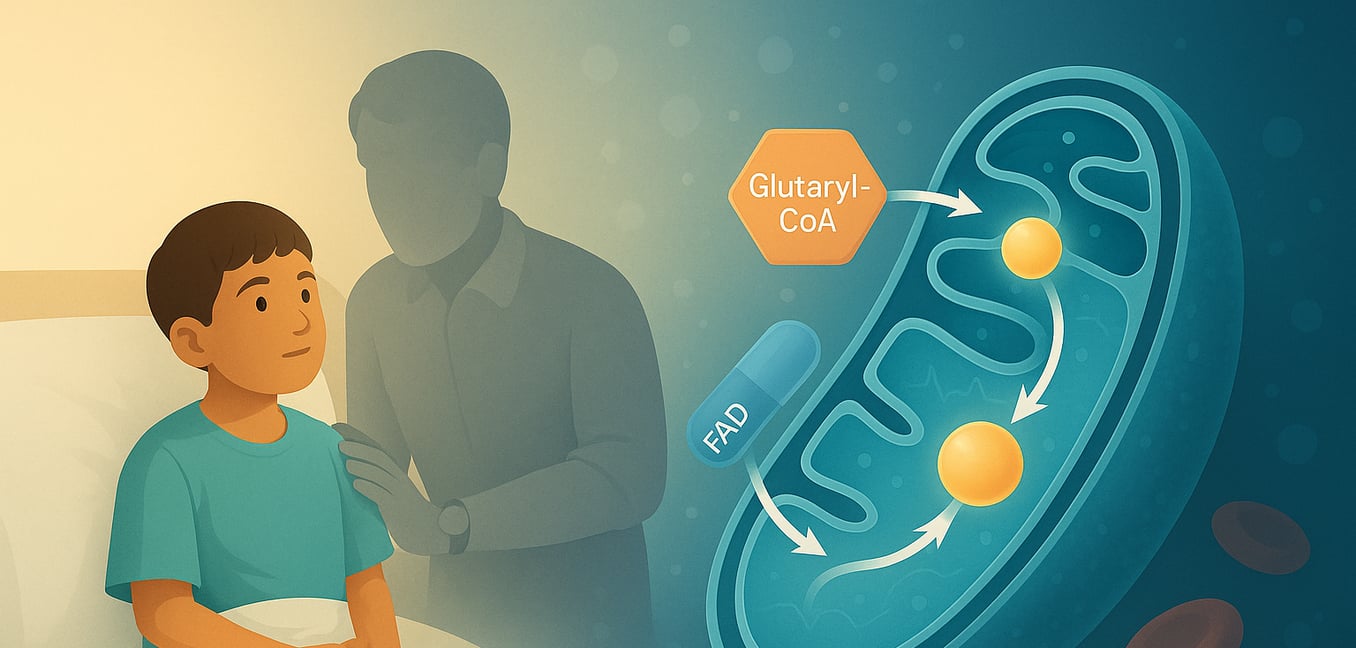



La glutaryl-CoA deidrogenasi (GCDH) è un enzima vitale situato all'interno dei mitocondri, le centrali energetiche delle nostre cellule. Codificato dal GCDH gene, questa proteina appartiene alla famiglia delle acil-CoA deidrogenasi (ACD) e svolge un ruolo fondamentale nei percorsi metabolici di degradazione di tre aminoacidi essenziali: L-lisina, L-idrossilisina e L-triptofano. Il corretto funzionamento della GCDH è cruciale per prevenire l'accumulo di sostanze potenzialmente dannose nel corpo. Quando questo enzima è carente o non funziona a causa di mutazioni genetiche, porta a un grave disturbo metabolico noto come acido glutammico tipo I (GA1), sottolineando l'importanza della sua attività per la salute complessiva, in particolare per lo sviluppo neurologico.
La funzione primaria della GCDH è di catalizzare una specifica reazione chimica: la decarbossilazione ossidativa della glutaryl-CoA in crotonil-CoA e anidride carbonica. Questo processo complesso avviene in diversi passaggi distinti all'interno della matrice mitocondriale, dove la GCDH esiste come un omotetramero (un complesso di quattro subunità identiche). La reazione inizia quando il substrato glutaryl-CoA si lega alla forma ossidata dell'enzima. Un residuo di aminoacidi cruciale nella GCDH, glutammato-370 (Glu370), funge da base catalitica, astraendo un protone (il protone alfa) dal substrato. Seguendo questo, un ione idruro viene trasferito dal beta-carbonio del substrato alla posizione N(5) del flavina adenina dinucleotide (FAD), un coenzima legato alla GCDH. Questo trasferimento riduce il FAD a FADH2. Questo passaggio facilita la decarbossilazione (rimozione di un gruppo carbossile come CO2) di un intermedio legato all'enzima, glutaconil-CoA, rompendo il suo legame Cγ-Cδ. Questa rottura porta alla formazione di un anione dienolato, un protone e anidride carbonica. L'intermedio dienolato viene quindi protonato, producendo il prodotto finale, crotonil-CoA, che viene successivamente rilasciato dal sito attivo dell'enzima insieme a CO2. Per completare il ciclo catalitico e rigenerare l'enzima per un altro turno, il FADH2 ridotto viene ri-ossidato a FAD trasferendo i suoi elettroni in due passaggi a singolo elettrone a un accettore di elettroni esterno, tipicamente la flavoproteina trasferente elettroni (ETF).
Comprendere la funzione della GCDH è fondamentale poiché il suo malfunzionamento ha gravi conseguenze. Negli individui con GA1, le mutazioni nel GCDH gene portano a una carenza di enzima GCDH funzionale. Di conseguenza, il corpo non può elaborare efficacemente la glutaryl-CoA. Questo blocco nel percorso metabolico causa l'accumulo di glutaryl-CoA e dei suoi metaboliti a monte. Questi vengono poi convertiti in altri composti, principalmente acido glutammico (GA), acido 3-idrossiglutarico (3-OH-GA) e, in misura minore, acido glutaconico, che si accumulano nei fluidi corporei e nei tessuti. Queste acidi organici accumulati sono neurotossici, particolarmente dannosi per specifiche aree del cervello, come i gangli basali, che sono cruciali per controllare il movimento. Questa neurotossicità è responsabile dei sintomi caratteristici del GA1, tra cui crisi encefalopatiche acute, distonia e altri gravi disturbi del movimento. L'accumulo di glutarylcarnitina (C5DC), un sottoprodotto non tossico, è anche una caratteristica e viene utilizzato nei test di screening neonatale per rilevare precocemente il disturbo, consentendo un intervento tempestivo per gestire la condizione, ad esempio, limitando la lisina nella dieta.
Che cos'è una carenza di deidrogenasi dell'idrossiacile a catena lunga?
La carenza di deidrogenasi dell'idrossiacile a catena lunga (LCHAD) è un disturbo metabolico ereditario che impedisce al corpo di degradare efficacemente determinati tipi di grassi, noti come acidi grassi a catena lunga, per convertirli in energia. Questa condizione si verifica a causa di una carenza o di un malfunzionamento dell'enzima LCHAD, che svolge un ruolo critico nel percorso di beta-ossidazione degli acidi grassi mitocondriali. Di conseguenza, questi acidi grassi a catena lunga non processati possono accumularsi nel corpo, portando a potenziali gravi conseguenze per la salute che interessano il fegato, il cuore, i muscoli e il cervello, particolarmente durante i periodi di digiuno o malattia. I sintomi chiave possono includere ipoglicemia (basso livello di zucchero nel sangue), debolezza muscolare (miopatia) e problemi cardiaci (cardiomiopatia), evidenziando la necessità di una diagnosi precoce e una gestione attenta, spesso coinvolgendo una dieta speciale.
Qual è la funzione del gene PKLR?
Il gene PKLR fornisce le istruzioni essenziali per sintetizzare l'enzima piruvato chinasi, che svolge un ruolo critico nella produzione di energia cellulare. Questo enzima è un attore chiave nella glicolisi, il percorso metabolico che degrada il glucosio (un tipo di zucchero) in piruvato, rilasciando energia nel processo. Il gene PKLR dirige specificamente la produzione di due forme di piruvato chinasi: il tipo L, prevalentemente attivo nel fegato, e il tipo R, che funziona nei globuli rossi. In entrambi i tessuti, la piruvato chinasi catalizza l'ultimo passaggio, ricco di energia, della glicolisi, facilitando il trasferimento di un gruppo fosfato dal fosfoenolpiruvato all'ADP per formare ATP, la principale moneta energetica della cellula.
