


Che cos'è la sindrome di Marfan?
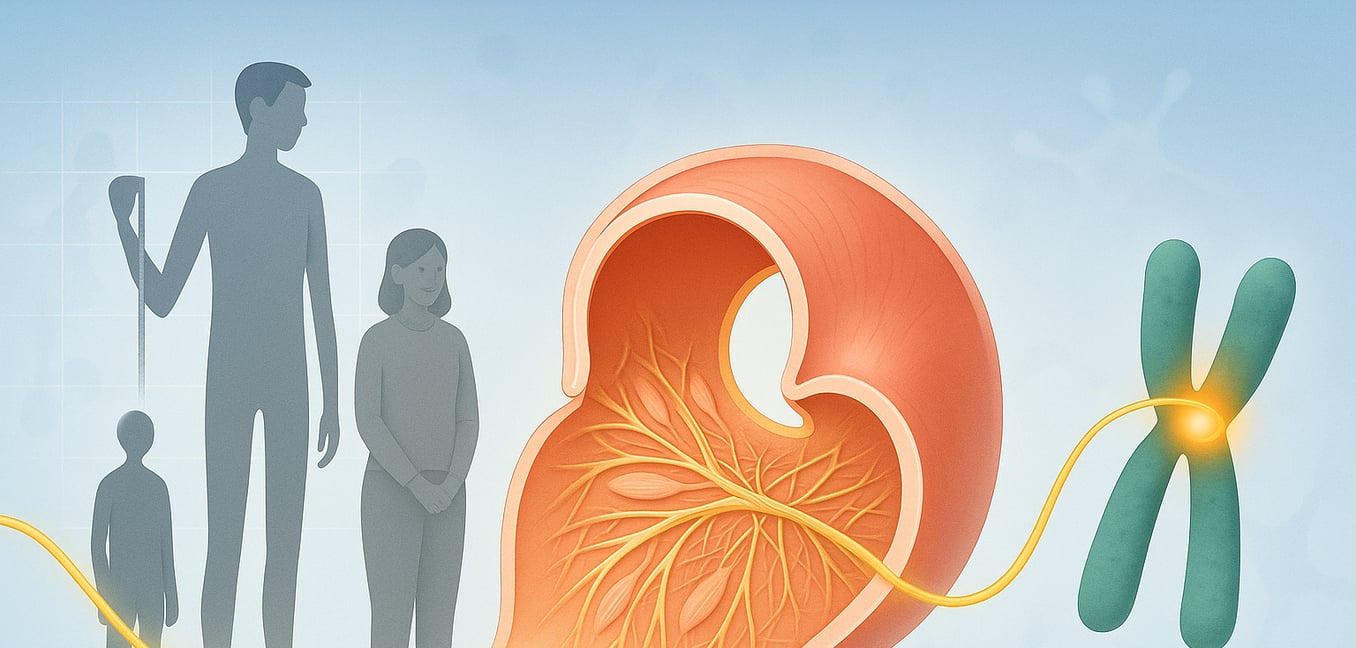
La sindrome di Marfan è un disturbo ereditario che colpisce il tessuto connettivo del corpo, il materiale fibroso che fornisce struttura e supporto agli organi, ai vasi sanguigni, alle ossa e ai muscoli. La condizione deriva da un difetto nel gene FBN1, responsabile della produzione della fibrillina-1, una proteina essenziale per la resistenza e l'elasticità del tessuto connettivo. Poiché questo tessuto è presente in tutto il corpo, la sindrome di Marfan può portare a una vasta gamma di sfide sanitarie, coinvolgendo più seriamente il cuore, lo scheletro e gli occhi.
I Due Percorsi della Sindrome di Marfan: Eredità e Mutazione Spontanea
Un individuo sviluppa la sindrome di Marfan attraverso uno dei due percorsi genetici. Comprendere questi due percorsi è il primo passo per apprezzare il ruolo centrale che gioca la storia familiare — e perché a volte è assente.
Il percorso più comune, che rappresenta circa il 75% dei casi, è l'ereditarietà diretta da un genitore. La condizione segue un modello autosomico dominante, il che significa che ereditare una sola copia del gene FBN1 mutato da un solo genitore è sufficiente per causare il disturbo. Questo dà a ciascun figlio di un genitore colpito una possibilità del 50% di sviluppare la sindrome di Marfan, una probabilità che si resetta con ogni gravidanza.
Il secondo percorso, responsabile del rimanente 25% dei casi, è una mutazione spontanea o "de novo". In questi casi, il cambiamento genetico nel gene FBN1 si verifica per la prima volta in un individuo, avvenendo casualmente nell'ovulo o nello spermatozoo di genitori non colpiti. Questo è un incidente biologico, non qualcosa che i genitori avrebbero potuto prevenire. Una volta che questa mutazione spontanea si verifica, tuttavia, l'individuo ha la sindrome di Marfan e può quindi trasmettere il gene alterato ai propri figli con la stessa probabilità del 50%.
Perché una Storia Familiare Dettagliata è Cruciale per la Diagnosi
Sebbene una diagnosi sia possibile senza di essa, una storia familiare completa è lo strumento più potente per identificare la sindrome di Marfan. Fornisce una mappa che può accorciare il percorso verso una diagnosi accurata e cure salvavita per diversi motivi chiave.
- Semplifica i criteri diagnostici. Le linee guida ufficiali, conosciute come criteri di Ghent, sono molto meno severe per gli individui con una storia familiare nota. Un singolo sintomo principale, come un aorta ingrandita, può essere sufficiente per confermare una diagnosi che altrimenti richiederebbe molte più prove.
- Abilita un trattamento più veloce. Abbassando la soglia diagnostica, una storia familiare consente una conferma più precoce. Questo significa che il monitoraggio cruciale del cuore e dell'aorta, così come i trattamenti preventivi, possono iniziare senza il ritardo causato dall'attesa di ulteriori sintomi.
- Allerta i parenti a rischio. Quando una persona viene diagnosticata, funge da allerta critica per altri membri della famiglia. Questo processo, chiamato screening a cascata, può identificare i parenti che portano il gene ma hanno sintomi lievi o assenti, lasciandoli ignari del loro rischio di una rottura aortica silenziosa ma mortale.
- Fornisce un contesto cruciale. Molte caratteristiche della sindrome di Marfan — come una statura alta, dita lunghe, piedi piatti o ipermobilità articolare — sono comuni nella popolazione generale. Di per sé, potrebbero non destare allerta. Tuttavia, quando un medico scopre che il padre di un paziente ha avuto un aneurisma aortico, queste caratteristiche sono improvvisamente viste come parte di un modello più ampio.
Quando la Storia Familiare è Complicata: Comprendere l'Espressività Variabile
Uno degli aspetti più perplessi della sindrome di Marfan è la sua "espressività variabile" — come può manifestarsi in modo così diverso da una persona all'altra, anche all'interno della stessa famiglia che condivide la stessa mutazione genetica. Un parente potrebbe avere gravi problemi cardiovascolari, mentre un altro ha solo lievi caratteristiche scheletriche e oculari. Questa variabilità clinica può complicare la diagnosi e sottolinea perché chiunque abbia una storia familiare debba essere valutato con attenzione.
La Mutazione Genica Specifica
Il gene FBN1 è grande, e la posizione precisa della mutazione su quel gene conta. Pensa al gene come a un lungo manuale di istruzioni; un errore di battitura in un capitolo critico può avere un effetto molto diverso rispetto a uno in una sezione meno vitale. Alcune mutazioni portano a una proteina strutturalmente difettosa che interferisce attivamente con la funzione normale, causando spesso malattie più gravi. Altre potrebbero semplicemente ridurre la quantità di proteina prodotta, portando a un diverso insieme di sfide.
L'Influenza dei Gen modulatori
Nessun gene funziona in isolamento. Ogni persona ha uno sfondo unico di migliaia di altri "geni modulatori" che possono influenzare la mutazione primaria FBN1. Questi geni agiscono come manopole di volume, aumentando o diminuendo la gravità di specifici sintomi. Possono influenzare quanto bene il corpo compensi per il tessuto connettivo difettoso o quanto rapidamente i tessuti si degradano. Questo background genetico aiuta a spiegare perché un fratello può sviluppare un'aorta che si ingrossa rapidamente mentre quella di un altro rimane stabile per anni.
Fattori Ambientali ed Epigenetici
Oltre al codice DNA, altri fattori contribuiscono alla variazione. Cambiamenti epigenetici — marcatori chimici che indicano ai nostri geni quando accendersi o spegnersi — possono differire tra i membri della famiglia e alterare come viene espresso il gene FBN1. Inoltre, fattori legati allo stile di vita e all'ambiente, come il controllo della pressione sanguigna e i livelli di stress fisico, pongono richieste uniche sul tessuto connettivo indebolito del corpo, portando a risultati sanitari diversi nel corso della vita.
