



Il viaggio con una malattia rara inizia spesso con una ricerca di risposte. Per le persone e le famiglie che navigano in una Malattia da Deposito Lisosomiale (LSD), quella ricerca ti porta nel mondo microscopico delle nostre cellule. Sappiamo che questo può essere un paesaggio intimidatorio da esplorare, pieno di termini scientifici complessi e profonda incertezza. Il nostro obiettivo è percorrere questo cammino con te, semplificando la scienza in informazioni chiare e di supporto. Crediamo nel potere della comprensione e il nostro impegno è fornire uno spazio in cui puoi trovare chiarezza, comunità e la rassicurazione che non sei solo in questo viaggio.
Che cos'è una Malattia da Deposito Lisosomiale?

Al cuore, una Malattia da Deposito Lisosomiale (LSD) è una sfida all'interno del “centro di riciclaggio” vitale della cellula, un organulo chiamato lisosoma. La storia delle LSD inizia con il lavoro pionieristico dello scienziato belga Christian de Duve, che scoprì il lisosoma negli anni '50, un traguardo che gli valse il Premio Nobel nel 1974. Identificò questi piccoli organuli come il sistema digestivo della cellula, pieni di proteine specializzate chiamate enzimi. In una cellula sana, i lisosomi sono pieni di questi enzimi che scompongono materiali complessi, come grassi e zuccheri, in componenti più semplici che la cellula può riutilizzare (Platt, 2018).
Per qualcuno con un LSD, una specifica mutazione genetica significa che uno di questi enzimi cruciali è assente o non funziona correttamente. La maggior parte delle LSD è autosomica recessiva, il che significa che un bambino deve ereditare una copia del gene mutato da entrambi i genitori per avere la condizione (Meikle et al., 1999).
Immagina il sistema di riciclaggio di una città in cui il team responsabile della lavorazione della plastica smette improvvisamente di funzionare. La plastica inizierebbe ad accumularsi, causando problemi in tutta la città. Questo è simile a ciò che accade all'interno delle cellule in un LSD. Il materiale specifico (chiamato substrato) che l'enzima assente doveva elaborare inizia ad accumularsi (Platt, 2018). Col passare del tempo, questo accumulo provoca l'ingrossamento dei lisosomi con questo materiale stoccato, che danneggia le cellule. Questa “crisi dei rifiuti cellulari” può portare alla vasta gamma di sintomi che colpiscono gli organi del corpo (Parenti, Andria, & Ballabio, 2015).
L'Odissea Diagnostica: uno Spettro Diversificato di Condizioni

I ricercatori hanno ora identificato oltre 70 tipi distinti di LSD. Anche se tutte originano da un problema all'interno del lisosoma, si manifestano in modi molto vari, ciascuna definita dall'enzima specifico che manca e dal substrato che di conseguenza si accumula. Alcune delle LSD più riconosciute includono:
Malattia di Gaucher: Causata da una carenza dell'enzima glucocerebrosidasi, che porta all'accumulo di una sostanza grassa chiamata glucocerebroside (Grabowski & Hopkin, 2022).
Malattia di Pompe: Coinvolge l'accumulo di uno zucchero complesso, il glicogeno, che impatta particolarmente le cellule muscolari a causa della mancanza dell'enzima acido α-glucosidasi (Kishnani et al., 2006).
Malattia di Fabry: Deriva da una carenza nell'enzima α-galattosidasi A, causando un tipo di grasso chiamato globotriaosylceramide che si accumula nelle cellule di tutto il corpo (Germain, 2010).
Mucopolisaccaridosi (MPS): Un gruppo di disturbi correlati (inclusi gli sindromi di Hurler, Hunter e Sanfilippo) in cui mancano diversi enzimi necessari per scomporre molecole complesse di zucchero chiamate glicosaminoglicani (GAG) (Parenti, Andria, & Ballabio, 2015).
Malattia di Niemann-Pick (Tipi A, B, C) & Malattia di Tay-Sachs: Altre LSD ben note che colpiscono la capacità del corpo di metabolizzare grassi (lipidi), portando a gravi danni neurologici (Platt, 2018).
Poiché i lisosomi sono presenti praticamente in ogni cellula, i sintomi delle LSD sono straordinariamente diversificati e progressivi. Possono influenzare il cervello e il sistema nervoso, il cuore, il sistema scheletrico, il fegato, la milza, i polmoni e persino gli occhi (Parenti, Andria, & Ballabio, 2015). Comprendiamo che ogni viaggio è diverso e la gravità e l'età di insorgenza possono variare drammaticamente, anche tra individui con la stessa condizione denominata.
Navigare nel Percorso verso una Risposta
Per molte famiglie, il percorso verso una diagnosi è un processo lungo e difficile, un'esperienza spesso chiamata “odissea diagnostica” (The LSD Collaborative, 2015). I primi sintomi possono essere sottili, non specifici e possono mimare malattie infantili più comuni, portando a ritardi frustranti e diagnosi errate (Wraith, 2002). Sappiamo quanto possa essere difficile questo periodo di incertezza, il che sottolinea la profonda sfida della consapevolezza tra coloro che non sono specializzati in queste condizioni rare. Uno studio ha rivelato che quasi l'89% dei medici di famiglia intervistati non aveva mai considerato un LSD come potenziale diagnosi per nessuno dei loro pazienti, il che sottolinea la profonda sfida della consapevolezza.
Il sollievo di ricevere finalmente una diagnosi—di avere un nome per i sintomi disorientanti e dolorosi—può essere immenso. Come ha condiviso un paziente, P., che vive con la malattia di Fabry, i suoi sintomi sono iniziati nell'infanzia, ma non è stata diagnosticata fino a decenni dopo, dopo che significativi sofferenze erano già avvenuti nella sua famiglia.
I metodi diagnostici sono diventati sempre più sofisticati. Di solito coinvolgono:
Saggetti Enzimatici: Esami del sangue che misurano il livello di attività di specifici enzimi lisosomiali. Un livello significativamente ridotto o assente è un indicatore chiave di un LSD (Sun, 2018).
Test Genetici: Confermano la diagnosi identificando la specifica mutazione nel gene responsabile dell'enzima, il che può talvolta aiutare a prevedere la gravità della malattia o guidare le scelte terapeutiche (Parenti, Andria, & Ballabio, 2015).
Screening Neonatale (NBS): Negli ultimi anni, un numero crescente di regioni ha aggiunto LSD trattabili come Pompe, Gaucher, Fabry e MPS I ai loro pannelli di screening neonatale standard (Burton, 2017). Identificando queste condizioni nei primi giorni di vita, prima che si verifichino danni significativi e irreversibili, il NBS offre la migliore opportunità possibile per cambiare il decorso della malattia.
Il Costo Umano: Più di un Semplice Grafico Medico
Sappiamo che l'impatto di un LSD si estende ben oltre i sintomi fisici. Queste sono condizioni croniche, progressive e spesso debilitanti che pongono un profondo onere medico, emotivo, sociale e finanziario su pazienti e le loro famiglie. Le realtà quotidiane possono includere la gestione del dolore cronico e della fatica debilitante. Ted, un uomo che vive con la malattia di Gaucher, ha descritto una fatica immobilizzante che era completamente diversa dalla stanchezza ordinaria, rendendo anche i compiti più semplici asfissiosi.
Il paesaggio emotivo è complesso, pieno dello stress di gestire una malattia cronica, l'incertezza del futuro e il dolore che può accompagnare il deterioramento della salute. Come ha condiviso Jennie, la madre di Sophia che ha la malattia di Pompe, il periodo dopo la diagnosi è stato terrificante: "La parte più spaventosa... era non avere risposte definite e non sapere cosa potrebbe succedere o quando." È un viaggio che rimodella la dinamica familiare e le scelte di vita, e ci impegniamo a fornire una comunità in cui queste esperienze profondamente personali possano essere condivise, validate e comprese.
Un'Idea diventa Realtà
Per decenni, le opzioni per trattare queste condizioni sono state tragicamente limitate. Ma un'idea rivoluzionaria, prima concepita nel 1964 dal scopritore del lisosoma, Christian de Duve, ha cambiato tutto (Beck, 2018). Il concetto era semplice ma profondo: e se potessimo sostituire l'enzima mancante dall'esterno? Questa intuizione ha posto le basi per la Terapia di Sostituzione Enzimatica (ERT) (de Duve, 1964).
Il viaggio dal concetto alla clinica è stato caratterizzato da una immensa dedizione scientifica. Negli anni '70, esperimenti cruciali dimostrarono che le cellule carenti in una piastra di laboratorio potevano assorbire enzimi mancanti secreti da cellule sane, un fenomeno chiamato "correzione incrociata." (Fratantoni, Hall, & Neufeld, 1968). Questo dimostrò che il principio di base poteva funzionare. Ulteriori ricerche identificarono l'"etichetta" cellulare che guida gli enzimi al lisosoma: una molecola di zucchero chiamata mannose-6-fosfato (M6P) che si lega a specifici recettori M6P sulla superficie della cellula (Kornfeld, 1986).
Il vero progresso clinico è stato guidato dal Dott. Roscoe Brady e dal suo team presso i National Institutes of Health (Beck, 2018). Concentrandosi sulla malattia di Gaucher, purificarono l'enzima necessario (glucocerebrosidasi) da placenta umana e lo modificarono biochimicamente per indirizzarlo verso le cellule giuste (Brady et al., 1974). I trial clinici furono un successo straordinario, portando alla prima approvazione da parte della FDA per un ERT, Ceredase™, nel 1991 (Beck, 2018). Questo ha segnato l'inizio di una nuova era.
Oggi, grazie alla tecnologia del DNA ricombinante, gli ERT non derivano più da tessuti umani. Vengono prodotti in modo sicuro e costante in grandi quantità in laboratori sicuri, un grande progresso che ha migliorato sicurezza e approvvigionamento (Platt, 2018).
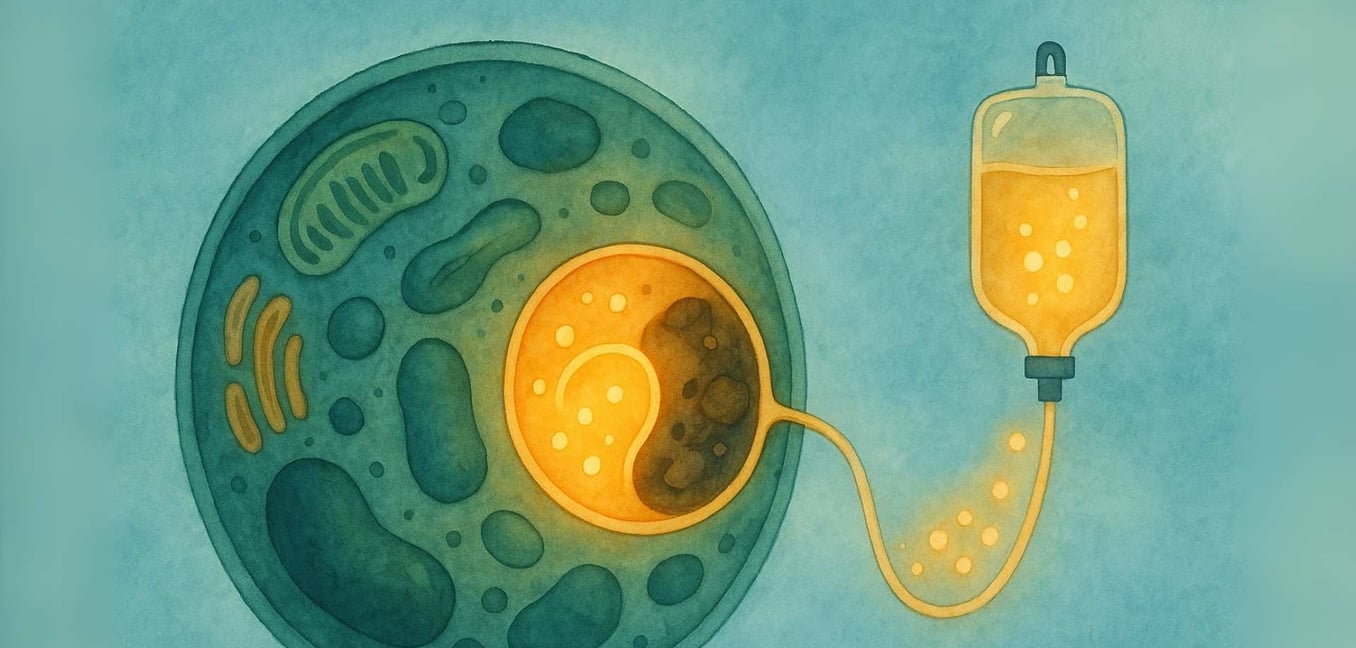
Come Funziona la Terapia di Sostituzione Enzimatica (ERT)

L'ERT è un trattamento medico progettato per fornire una versione funzionale dell'enzima mancante direttamente alle cellule che ne hanno più bisogno. Ecco uno sguardo più da vicino al processo:
L'Infusione: La terapia viene somministrata per via endovenosa (IV), tipicamente in un'infusione lenta nel corso di diverse ore, ogni una o due settimane (Beck, 2018).
Il Sistema di Consegna: Questi enzimi sono ingegnerizzati con quell'"etichette" M6P speciale che le cellule del corpo possono riconoscere. Questo consente all'enzima di legarsi ai recettori M6P sulla superficie cellulare, agendo come una chiave che si inserisce in una serratura specifica (Kornfeld, 1986).
Azione Cellulare: Una volta che la cellula riconosce e si lega all'enzima, lo accoglie all'interno attraverso un processo chiamato endocitosi e lo guida direttamente verso i lisosomi. Lì, l’enzima sostitutivo può finalmente iniziare a lavorare, scomponendo i materiali accumulati e aiutando a ripristinare un equilibrio più sano all'interno della cellula (Kornfeld, 1986).
Il Percorso del Trattamento: Speranza e Aspettative Oneste
L'ERT è un trattamento, non una cura (Platt, 2018). Non corregge il difetto genetico sottostante e richiede un impegno a vita per infusione regolare, una parte significativa della vita per i pazienti e le loro famiglie (Beck, 2018).
Inoltre, l'ERT affronta diversi ostacoli significativi:
La Barriera Emato-Encefalica (BBB): Questa è senza dubbio la sfida più grande. La BBB è uno scudo protettivo che impedisce a grandi molecole, inclusi gli enzimi ERT, di entrare nel cervello. Poiché si stima che due terzi delle LSD abbiano sintomi neurologici, l'ERT standard spesso non riesce ad affrontare il declino cognitivo o altri problemi del SNC, anche se aiuta il resto del corpo.
La Risposta Immunitaria: Poiché l'enzima infuso è una proteina, il corpo può talvolta vederlo come “estraneo” e sviluppare anticorpi anti-farmaco (ADA). Questi anticorpi possono, in alcuni casi, ridurre l'efficacia della terapia o causare reazioni associate all'infusione (IAR), come febbre, eruzione cutanea o reazioni allergiche più gravi.
Consegna a "Siti Santuario": Oltre al cervello, alcuni tessuti sono difficili da raggiungere per le grandi molecole enzimatiche. Questi includono ossa dense, cartilagini avascolari e valvole cardiache, il che significa che problemi scheletrici e malattie valvolari cardiache possono progredire nonostante il trattamento.
Espandere l'Arsenale di Terapie

Le limitazioni dell'ERT hanno ispirato i ricercatori e la nostra comunità a spingere incessantemente per nuove e migliori terapie. Il futuro del trattamento è focalizzato su un attacco a più fronti a queste condizioni, con diverse strategie entusiasmanti che stanno emergendo:
Terapia di Riduzione del Substrato (SRT): Invece di sostituire l'enzima, questo approccio utilizza un farmaco orale per inibire un enzima coinvolto nella produzione di substrato in primo luogo. Questo “alleggerisce” efficacemente il carico sui lisosomi compromessi. Farmaci approvati come Miglustat e Eliglustat sono usati per la malattia di Gaucher di tipo 1.
Terapia con Chaperone Farmacologico (PCT): Per pazienti il cui corpo produce un enzima che è mal ripiegato ma ha ancora una certa funzione, questi piccoli farmaci orali agiscono come un “supporto”. Si legano all'enzima instabile, aiutando a piegarlo correttamente in modo che possa superare il controllo di qualità della cellula e raggiungere il lisosoma per svolgere la sua funzione. Migalastat è un chaperone approvato per pazienti con malattia di Fabry con specifiche mutazioni “ammissibili”.
Trapianto di Cellule Staminali Ematopoietiche (HSCT): Per alcune LSD gravi con coinvolgimento del SNC, come la MPS I (sindrome di Hurler), l'HSCT (o trapianto di midollo osseo) da un donatore sano può essere un'opzione. L'idea è che le cellule derivate dal donatore viaggino verso il cervello e forniscano una fonte locale e continua dell'enzima mancante. Tuttavia, questa è una procedura ad alto rischio con potenziali complicazioni significative.
Terapia Genica: Questa è una delle frontiere più entusiasmanti e potenzialmente trasformative. L'obiettivo è fornire una copia funzionale del gene corretto nelle cellule di un paziente, consentendo loro di produrre l'enzima da soli. Utilizzando vettori virali ingegnerizzati o altri sistemi di somministrazione, i ricercatori stanno esplorando modi per fornire un trattamento duraturo, potenzialmente una tantum, che corregga la causa sottostante della malattia. Sono in corso trial clinici per Pompe, Fabry e diversi disturbi MPS, offrendo una potente fonte di speranza.
Avanzare Insieme
Il percorso dalla scoperta fondamentale del lisosoma allo sviluppo dell'ERT e all'alba della medicina basata sui geni è una testimonianza del potere dell'indagine scientifica e della resilienza umana. Ci impegniamo a continuare questa crescente marcia, insieme. Ci impegneremo sempre a fornire informazioni chiare e affidabili, a coltivare una comunità di supporto e cura e a promuovere la ricerca che porta speranza tangibile per un futuro più luminoso.
Per una panoramica concisa ma perspicace della Terapia di Sostituzione Enzimatica, ti invitiamo a sintonizzarti sul nostro nuovo episodio di podcast. È progettato per semplificare questo argomento complesso in segmenti facilmente digeribili.
<iframe width="560" height="315" src="https://www.youtube.com/embed/T91yim2x8ac?si=5aRF203Rw9fqjAoN" title="Riproduttore video YouTube" frameborder="0" allow="accelerometer; autoplay; clipboard-write; encrypted-media; gyroscope; picture-in-picture; web-share" referrerpolicy="strict-origin-when-cross-origin" allowfullscreen></iframe>
Riferimenti
Nobel Prize Outreach AB. (2024). Il Premio Nobel in Fisiologia o Medicina 1974. NobelPrize.org.
de Duve, C. (2013). Christian de Duve: Esploratore della cellula che ha scoperto nuovi organuli utilizzando una centrifuga. Proceedings of the National Academy of Sciences, 110(31), 12559-12561.
La Rockefeller University. (n.d.). "Esplorare le cellule con una centrifuga": La scoperta del lisosoma. Ospedale Centennial.
MSD Manual Professional Edition. (n.d.). Panoramica dei Disturbi da Deposito Lisosomiale.
Columbia University Irving Medical Center. (n.d.). Disturbi da Deposito Lisosomiale Pediatrici.
Cleveland Clinic. (2022, 27 Giugno). Malattie e Disturbi Lisosomiali.
Cleveland Clinic. (2022, 27 Giugno). Malattie e Disturbi Lisosomiali.
Bioscience Institute. (n.d.). Malattie da Deposito Lisosomiali (LSDS).
van der Meijden, J. C., et al. (2010). 'Dottor Google' che termina l'odissea diagnostica nei disturbi da deposito lisosomiale. Archives of Disease in Childhood, 95(8), 642-644.
Medicover Genetics. (2025, 26 Febbraio). L'odissea diagnostica: La ricerca di risposte per le malattie rare.
Rare Diseases South Africa. (2023, 28 Febbraio). Affrontare le sfide della diagnosi e del trattamento delle malattie da deposito lisosomiale.
Greenwood Genetic Center. (2024, 14 Agosto). Disturbi da Deposito Lisosomiale: Malattia di Gaucher e di Fabry.
Wikipedia. (n.d.). Terapia di sostituzione enzimatica.
Beck, M. (2018). Terapia di sostituzione enzimatica e oltre—In memoria di Roscoe O. Brady, M.D. (1923–2016). Journal of Inherited Metabolic Disease, 41(1), 3-13.
Park, J. J., & Lee, K. (2022). Glicosilazione della mannose-6-fosfato per il targeting lisosomiale: varie applicazioni dalla terapia di sostituzione enzimatica ai chimeri mirati ai lisosomi. Animal Cells and Systems, 26(3), 84-91.
U.S. Food & Drug Administration (FDA). (1991). Cerca designazioni e approvazioni di farmaci orfani: Ceredase.
National Gaucher Foundation. (n.d.). Il 25° Anniversario dell'Approvazione della FDA per l'ERT.
Begley, D. J. (2015). Modificare il trasporto attraverso la barriera emato-encefalica per portare speranza ai pazienti con malattie da deposito lisosomiale. Journal of Cerebral Blood Flow & Metabolism, 35(1), 3-5.
Pardridge, W. M. (2015). Targeting della barriera emato-encefalica degli enzimi lisosomiali terapeutici. Disturbi da Deposito Lisosomiale, 327-343.
Aflaki, E., et al. (2018). Terapie di sostituzione enzimatica: qual è la migliore opzione?. Current Pharmaceutical Design, 24(11), 1238-1250.
Giugliani, R., et al. (2018). Terapia di sostituzione enzimatica per mucopolisaccaridosi: nuovi sviluppi e risultati clinici. Expert Opinion on Orphan Drugs, 6(4), 277-287.
Gaucher Disease News. (n.d.). Terapia di riduzione del substrato per la malattia di Gaucher.
National Gaucher Foundation. (n.d.). Terapia di Riduzione del Substrato.
Shin, S-H., et al. (2007). Chaperone farmacologico che corregge il deposito lisosomiale nella malattia di Fabry causata da varianti non competenti nel traffico. American Journal of Physiology-Cell Physiology, 292(5), C1879-C1887.
Fabry Disease News. (2024, 19 Aprile). Terapia con chaperone per la malattia di Fabry.
National MPS Society. (n.d.). HSCT.
La-Fauci, G., & A.H. Schuchman. (2016). Terapia genica per i disturbi da deposito lisosomiale: recenti progressi e limitazioni. Journal of Inborn Errors of Metabolism and Screening, 4, 1-7.
Iimori, T., et al. (2023). Terapia genica per malattie da deposito lisosomiale: prospettive attuali degli studi clinici. Frontiers in Genetics, 14, 1064924.
Platt, F. M., d'Azzo, A., Davidson, B. L., Neufeld, E. F., & Tifft, C. J. (2018). Malattie da Deposito Lisosomiale. Nature Reviews Disease Primers, 4(1), 27.