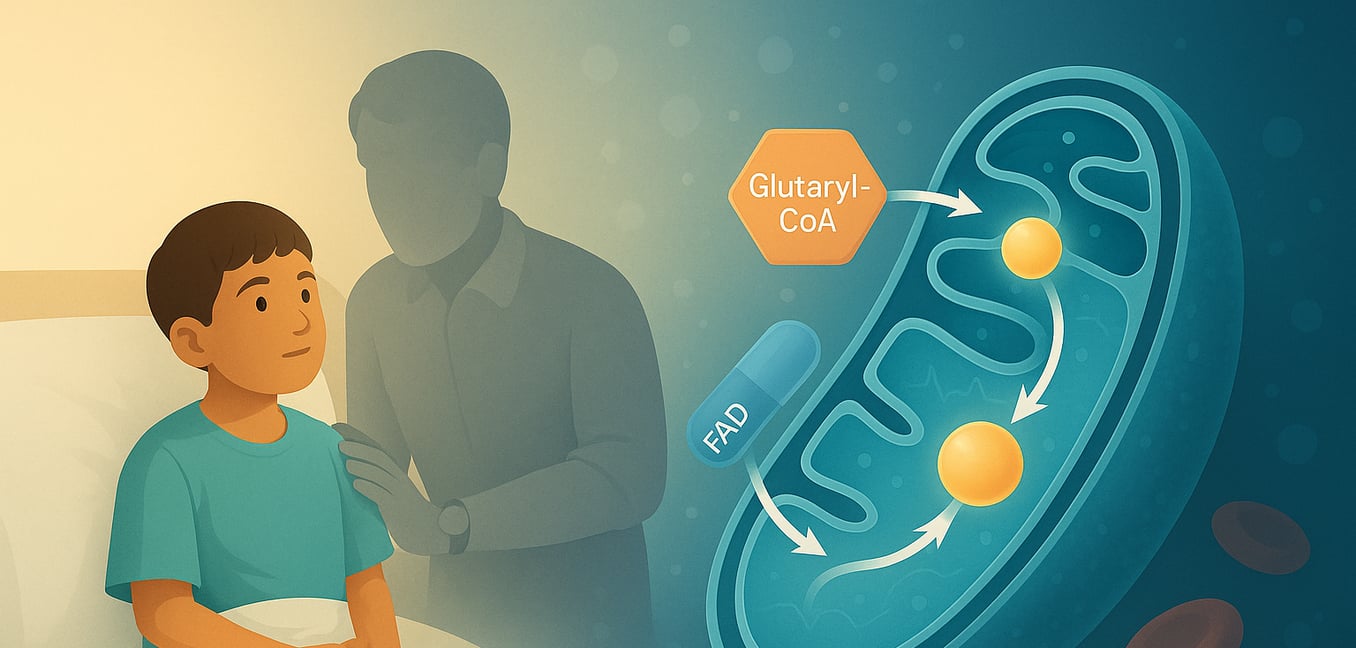



Glutaryl-CoA dehydrogenase (GCDH) is a vital enzyme located within the mitochondria, the powerhouses of our cells. Encoded by the GCDH gene, this protein belongs to the acyl-CoA dehydrogenase (ACD) family and plays a critical role in the metabolic breakdown pathways of three essential amino acids: L-lysine, L-hydroxylysine, and L-tryptophan. The proper functioning of GCDH is crucial for preventing the buildup of potentially harmful substances in the body. When this enzyme is deficient or malfunctioning due to genetic mutations, it leads to a serious metabolic disorder known as glutaric aciduria type I (GA1), underscoring the importance of its activity for overall health, particularly neurological development.
The primary function of GCDH is to catalyze a specific chemical reaction: the oxidative decarboxylation of glutaryl-CoA into crotonyl-CoA and carbon dioxide. This complex process occurs in several distinct steps within the mitochondrial matrix, where GCDH exists as a homotetramer (a complex of four identical subunits). The reaction begins when the glutaryl-CoA substrate binds to the oxidized form of the enzyme. A crucial amino acid residue in GCDH, glutamate-370 (Glu370), acts as a catalytic base, abstracting a proton (the alpha-proton) from the substrate. Following this, a hydride ion is transferred from the beta-carbon of the substrate to the N(5) position of flavin adenine dinucleotide (FAD), a coenzyme bound to GCDH. This transfer reduces FAD to FADH2. This step facilitates the decarboxylation (removal of a carboxyl group as CO2) of an enzyme-bound intermediate, glutaconyl-CoA, by breaking its Cγ-Cδ bond. This breakage results in the formation of a dienolate anion, a proton, and carbon dioxide. The dienolate intermediate is then protonated, yielding the final product, crotonyl-CoA, which is subsequently released from the enzyme's active site along with CO2. To complete the catalytic cycle and regenerate the enzyme for another round, the reduced FADH2 is re-oxidized back to FAD by transferring its electrons in two single-electron steps to an external electron acceptor, typically the electron-transferring flavoprotein (ETF).
Understanding the function of GCDH is paramount because its impairment has severe consequences. In individuals with GA1, mutations in the GCDH gene lead to a deficiency in functional GCDH enzyme. As a result, the body cannot effectively process glutaryl-CoA. This blockage in the metabolic pathway causes glutaryl-CoA and its upstream metabolites to accumulate. These are then converted into other compounds, primarily glutaric acid (GA), 3-hydroxyglutaric acid (3-OH-GA), and to a lesser extent, glutaconic acid, which build up in body fluids and tissues. These accumulated organic acids are neurotoxic, particularly damaging to specific regions of the brain like the basal ganglia, which are crucial for controlling movement. This neurotoxicity is responsible for the characteristic symptoms of GA1, including acute encephalopathic crises, dystonia, and other severe movement disorders. The accumulation of glutarylcarnitine (C5DC), a non-toxic byproduct, is also a hallmark and is used in newborn screening tests to detect the disorder early, allowing for timely intervention to manage the condition by, for example, restricting dietary lysine.
What is a long-chain hydroxyacyl CoA dehydrogenase deficiency?
Long-chain hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency is an inherited metabolic disorder that prevents the body from effectively breaking down certain types of fats, known as long-chain fatty acids, to convert them into energy. This condition occurs due to a deficiency or malfunction of the LCHAD enzyme, which plays a critical role in the mitochondrial fatty acid beta-oxidation pathway. As a result, these unprocessed long-chain fatty acids can accumulate in the body, leading to potentially serious health issues affecting the liver, heart, muscles, and brain, particularly during periods of fasting or illness. Key symptoms can include low blood sugar (hypoglycemia), muscle weakness (myopathy), and heart problems (cardiomyopathy), highlighting the need for early diagnosis and careful management, often involving a special diet.
What is the function of the PKLR gene?
The PKLR gene provides the essential instructions for synthesizing the enzyme pyruvate kinase, which plays a critical role in cellular energy production. This enzyme is a key player in glycolysis, the metabolic pathway that breaks down glucose (a type of sugar) into pyruvate, releasing energy in the process. The PKLR gene specifically directs the production of two forms of pyruvate kinase: the L-type, predominantly active in the liver, and the R-type, which functions in red blood cells. In both tissues, pyruvate kinase catalyzes the final, energy-yielding step of glycolysis, facilitating the transfer of a phosphate group from phosphoenolpyruvate to ADP to form ATP, the cell's primary energy currency.
