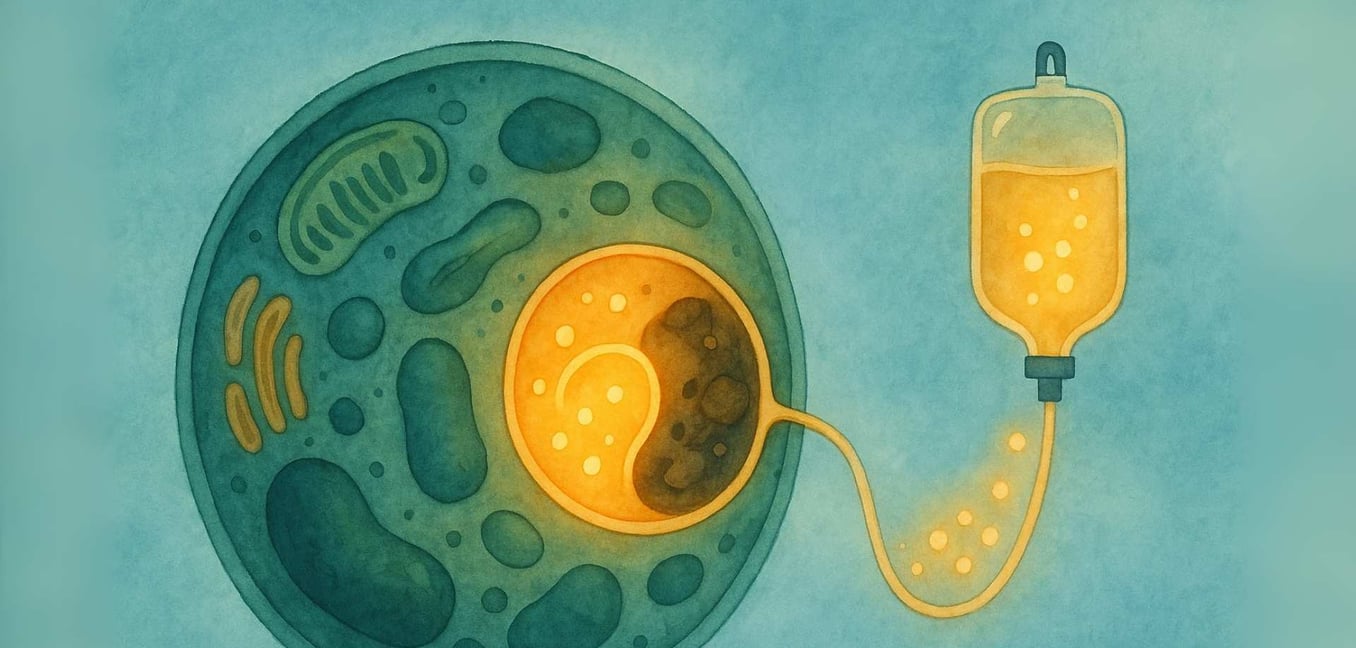



The journey with a rare disease often begins with a search for answers. For individuals and families navigating a Lysosomal Storage Disease (LSD), that search takes you into the microscopic world of our own cells. We know that this can be an intimidating landscape to explore, filled with complex scientific terms and profound uncertainty. Our goal is to walk this path with you, breaking down the science into clear, supportive information. We believe in the power of understanding, and our commitment is to provide a space where you can find clarity, community, and the reassurance that you are not alone on this journey.
What is a Lysosomal Storage Disease?

At its heart, a Lysosomal Storage Disease (LSD) is a challenge within the cell’s vital “recycling center,” an organelle called the lysosome. The story of LSDs begins with the groundbreaking work of Belgian scientist Christian de Duve, who discovered the lysosome in the 1950s—an achievement that earned him a Nobel Prize in 1974. He identified these tiny organelles as the cell's digestive system, filled with specialized proteins called enzymes. In a healthy cell, lysosomes are filled with these enzymes that break down complex materials, like fats and sugars, into simpler components the cell can reuse (Platt, 2018).
For someone with an LSD, a specific gene mutation means one of these crucial enzymes is either missing or doesn’t work correctly. Most LSDs are autosomal recessive, meaning a child must inherit a copy of the mutated gene from both parents to have the condition (Meikle et al., 1999).
Imagine a city’s recycling system where the team responsible for processing plastic suddenly stops working. The plastic would begin to pile up, causing problems across the city. This is similar to what happens inside the cells in an LSD. The specific material (called a substrate) that the missing enzyme was meant to process begins to accumulate (Platt, 2018). Over time, this buildup causes the lysosomes to become engorged with this stored material, which damages the cells. This "cellular garbage crisis" can eventually lead to the wide range of symptoms that affect organs throughout the body (Parenti, Andria, & Ballabio, 2015).
The Diagnostic Odyssey: A Diverse Spectrum of Conditions

Researchers have now identified over 70 distinct types of LSDs. While they all originate from a problem within the lysosome, they manifest in a wide variety of ways, each defined by the specific enzyme that is lacking and the substrate that consequently builds up. Some of the more widely recognized LSDs include:
-
Gaucher Disease: Caused by a deficiency of the enzyme glucocerebrosidase, leading to the accumulation of a fatty substance called glucocerebroside (Grabowski & Hopkin, 2022).
-
Pompe Disease: Involves the accumulation of a complex sugar, glycogen, particularly impacting muscle cells due to a lack of the enzyme acid α-glucosidase (Kishnani et al., 2006).
-
Fabry Disease: Results from a deficiency in the α-galactosidase A enzyme, causing a type of fat called globotriaosylceramide to build up in cells throughout the body (Germain, 2010).
-
Mucopolysaccharidoses (MPS): A group of related disorders (including Hurler, Hunter, and Sanfilippo syndromes) where different enzymes needed to break down complex sugar molecules called glycosaminoglycans (GAGs) are missing (Parenti, Andria, & Ballabio, 2015).
-
Niemann-Pick Disease (Types A, B, C) & Tay-Sachs Disease: Other well-known LSDs that affect the body’s ability to metabolize fats (lipids), leading to severe neurological damage (Platt, 2018).
Because lysosomes are present in virtually every cell, the symptoms of LSDs are extraordinarily diverse and progressive. They can impact the brain and nervous system, the heart, the skeletal system, the liver, spleen, lungs, and even the eyes (Parenti, Andria, & Ballabio, 2015). We understand that every journey is different, and the severity and age of onset can vary dramatically, even among individuals with the same named condition.
Navigating the Path to an Answer
For many families, the path to a diagnosis is a long and arduous process, an experience often called a "diagnostic odyssey" (The LSD Collaborative, 2015). Early symptoms can be subtle, non-specific, and may mimic more common childhood illnesses, leading to frustrating delays and misdiagnoses (Wraith, 2002). We know how difficult this period of uncertainty can be, which underscores the profound challenge of awareness among those not specialized in these rare conditions. One study revealed that nearly 89% of surveyed primary care physicians had never even considered an LSD as a potential diagnosis for any of their their patients, which underscores the profound challenge of awareness.
The relief of finally receiving a diagnosis—of having a name for the perplexing and painful symptoms—can be immense. As one patient, P., who lives with Fabry disease, shared, her symptoms began in childhood, but she was not diagnosed until decades later, after significant suffering had already occurred in her family.
Diagnostic methods have become increasingly sophisticated. They typically involve:
-
Enzyme Assays: Blood tests that measure the activity level of specific lysosomal enzymes. A significantly reduced or absent level is a key indicator of an LSD (Sun, 2018).
-
Genetic Testing: Confirms the diagnosis by identifying the specific mutation in the gene responsible for the enzyme, which can sometimes help predict disease severity or guide treatment choices (Parenti, Andria, & Ballabio, 2015).
-
Newborn Screening (NBS): In recent years, a growing number of regions have added treatable LSDs like Pompe, Gaucher, Fabry, and MPS I to their standard newborn screening panels (Burton, 2017). By identifying these conditions in the first days of life, before significant and irreversible damage can occur, NBS offers the best possible opportunity to change the course of the disease.
The Human Cost: More Than Just a Medical Chart
We know that the impact of an LSD extends far beyond the physical symptoms. These are chronic, progressive, and often debilitating conditions that place a profound medical, emotional, social, and financial burden on patients and their their families. The daily realities can include managing chronic pain and debilitating fatigue. Ted, a man living with Gaucher disease, described an immobilizing fatigue that was entirely different from ordinary tiredness, making even simple tasks feel monumental.
The emotional landscape is complex, filled with the stress of managing a chronic illness, the uncertainty of the future, and the grief that can accompany declining health. As Jennie, the mother of Sophia who has Pompe disease, shared, the time after diagnosis was terrifying: "The scariest part...was not having definite answers and not knowing what could happen or when." It's a journey that reshapes family dynamics and life choices, and we are committed to providing a community where these deeply personal experiences can be shared, validated, and understood.
An Idea Becomes Reality
For decades, options for treating these conditions were heartbreakingly limited. But a revolutionary idea, first conceptualized in 1964 by the lysosome's discoverer, Christian de Duve, changed everything (Beck, 2018). The concept was simple yet profound: what if we could replace the missing enzyme from the outside? This insight laid the foundation for Enzyme Replacement Therapy (ERT) (de Duve, 1964).
The journey from concept to clinic was one of immense scientific dedication. In the 1970s, crucial experiments showed that deficient cells in a lab dish could absorb missing enzymes secreted by healthy cells, a phenomenon called "cross-correction." (Fratantoni, Hall, & Neufeld, 1968). This proved the basic principle could work. Further research identified the cellular "address label" that guides enzymes to the lysosome: a sugar molecule called mannose-6-phosphate (M6P) that binds to specific M6P receptors on the cell surface (Kornfeld, 1986).
The true clinical breakthrough was spearheaded by Dr. Roscoe Brady and his team at the National Institutes of Health (Beck, 2018). Focusing on Gaucher disease, they purified the needed enzyme (glucocerebrosidase) from human placentas and biochemically modified it to target the right cells (Brady et al., 1974). Clinical trials were a stunning success, leading to the first FDA approval for an ERT, Ceredase™, in 1991 (Beck, 2018). This heralded a new era.
Today, thanks to recombinant DNA technology, ERTs are no longer derived from human tissue. They are safely and consistently produced in large quantities in secure laboratory settings, a major advance that has improved safety and supply (Platt, 2018).
How Enzyme Replacement Therapy (ERT) Works

ERT is a medical treatment designed to deliver a functional version of the missing enzyme directly to the cells that need it most. Here’s a closer look at the process:
-
The Infusion: The therapy is administered intravenously (IV), typically in a slow infusion over several hours, every one to two weeks (Beck, 2018).
-
The Delivery System: These enzymes are engineered with that special M6P "address label" that the body’s cells can recognize. This allows the enzyme to bind to M6P receptors on the cell surface, acting like a key fitting into a specific lock (Kornfeld, 1986).
-
Cellular Action: Once the cell recognizes and binds to the enzyme, it welcomes it inside through a process called endocytosis and guides it directly to the lysosomes. There, the replacement enzyme can finally get to work, breaking down the accumulated materials and helping to restore a healthier balance within the cell (Kornfeld, 1986).
The Treatment Journey: Hope and Honest Expectations
ERT is a treatment, not a cure (Platt, 2018). It does not correct the underlying genetic defect and requires a lifelong commitment to regular infusions, a significant part of life for patients and their families (Beck, 2018).
Furthermore, ERT faces several significant hurdles:
-
The Blood-Brain Barrier (BBB): This is arguably the biggest challenge. The BBB is a protective shield that prevents large molecules, including ERT enzymes, from entering the brain. Since an estimated two-thirds of LSDs have neurological symptoms, standard ERT often fails to address cognitive decline or other CNS issues, even as it helps the rest of the body.
-
The Immune Response: Because the infused enzyme is a protein, the body can sometimes see it as "foreign" and develop anti-drug antibodies (ADAs). These antibodies can, in some cases, reduce the effectiveness of the therapy or cause infusion-associated reactions (IARs), like fever, rash, or more severe allergic responses.
-
Delivery to "Sanctuary Sites": Beyond the brain, certain tissues are difficult for the large enzyme molecules to reach. These include dense bone, avascular cartilage, and heart valves, meaning skeletal issues and valvular heart disease may progress despite treatment.
Expanding the Arsenal of Therapies

The limitations of ERT have inspired researchers and our community to push relentlessly for new and better therapies. The future of treatment is focused on a multi-pronged attack on these conditions, with several exciting strategies emerging:
-
Substrate Reduction Therapy (SRT): Instead of replacing the enzyme, this approach uses an oral medication to inhibit an enzyme involved in producing the substrate in the first place. This effectively "lightens the load" on the compromised lysosomes. Approved drugs like Miglustat and Eliglustat are used for Gaucher disease type 1.
-
Pharmacological Chaperone Therapy (PCT): For patients whose bodies produce an enzyme that is misfolded but still has some potential function, these small oral medicines act as a "scaffold." They bind to the unstable enzyme, helping it fold correctly so it can pass the cell's quality control and get to the lysosome to do its job. Migalastat is an approved chaperone for Fabry patients with specific "amenable" mutations.
-
Hematopoietic Stem Cell Transplantation (HSCT): For some severe LSDs with CNS involvement, like MPS I (Hurler syndrome), HSCT (or bone marrow transplant) from a healthy donor can be an option. The idea is that donor-derived cells will travel to the brain and provide a local, continuous source of the missing enzyme. However, this is a very high-risk procedure with significant potential complications.
-
Gene Therapy: This is one of the most exciting and potentially transformative frontiers. The goal is to deliver a functional copy of the correct gene into a patient's own cells, enabling them to produce the enzyme themselves. Using engineered viral vectors or other delivery systems, researchers are exploring ways to provide a long-lasting, potentially one-time treatment that corrects the root cause of the disease. Clinical trials are underway for Pompe, Fabry, and several MPS disorders, offering a powerful source of hope.
Marching Forward, Together
The path from the fundamental discovery of the lysosome to the development of ERT and the dawn of gene-based medicine is a testament to the power of scientific inquiry and human resilience. We are committed to continuing this forward march, together. We will always strive to provide trustworthy, clear information, to foster a supportive and caring community, and to champion the research that brings tangible hope for a brighter future.
For a concise yet insightful overview of Enzyme Replacement Therapy, we invite you to tune into our new podcast episode. It's designed to break down this complex topic into easily digestible segments.
https://youtu.be/T91yim2x8ac?si=Rla7FYMAWxCLUVc8
References
Nobel Prize Outreach AB. (2024). The Nobel Prize in Physiology or Medicine 1974. NobelPrize.org.
de Duve, C. (2013). Christian de Duve: Explorer of the cell who discovered new organelles by using a centrifuge. Proceedings of the National Academy of Sciences, 110(31), 12559-12561.
The Rockefeller University. (n.d.). "Exploring Cells With a Centrifuge": The Discovery of the Lysosome. Hospital Centennial.
MSD Manual Professional Edition. (n.d.). Overview of Lysosomal Storage Disorders.
Columbia University Irving Medical Center. (n.d.). Pediatric Lysosomal Storage Disorders.
Cleveland Clinic. (2022, June 27). Lysosomal Storage Diseases & Disorders.
Cleveland Clinic. (2022, June 27). Lysosomal Storage Diseases & Disorders.
Bioscience Institute. (n.d.). Lysosomal Storage Diseases (LSDS).
van der Meijden, J. C., et al. (2010). 'Doctor Google' ending the diagnostic odyssey in lysosomal storage disorders. Archives of Disease in Childhood, 95(8), 642-644.
Medicover Genetics. (2025, February 26). The diagnostic odyssey: The search for answers for rare diseases.
Rare Diseases South Africa. (2023, February 28). Tackling the challenges of lysosomal storage diseases diagnosis and treatment.
Greenwood Genetic Center. (2024, August 14). Lysosomal Storage Disorders: Gaucher and Fabry Disease.
Wikipedia. (n.d.). Enzyme replacement therapy.
Beck, M. (2018). Enzyme replacement therapy and beyond—in memoriam Roscoe O. Brady, M.D. (1923–2016). Journal of Inherited Metabolic Disease, 41(1), 3-13.
Park, J. J., & Lee, K. (2022). Mannose-6-phosphate glycan for lysosomal targeting: various applications from enzyme replacement therapy to lysosome-targeting chimeras. Animal Cells and Systems, 26(3), 84-91.
U.S. Food & Drug Administration (FDA). (1991). Search Orphan Drug Designations and Approvals: Ceredase.
National Gaucher Foundation. (n.d.). The 25th Anniversary of FDA Approval of ERT.
Begley, D. J. (2015). Modifying blood–brain barrier transport to bring hope for patients with lysosomal storage diseases. Journal of Cerebral Blood Flow & Metabolism, 35(1), 3-5.
Pardridge, W. M. (2015). Blood–Brain Barrier Targeting of Therapeutic Lysosomal Enzymes. Lysosomal Storage Disorders, 327-343.
Aflaki, E., et al. (2018). Enzyme replacement therapies: what is the best option?. Current Pharmaceutical Design, 24(11), 1238-1250.
Giugliani, R., et al. (2018). Enzyme replacement therapy for mucopolysaccharidoses: new developments and clinical outcomes. Expert Opinion on Orphan Drugs, 6(4), 277-287.
Gaucher Disease News. (n.d.). Substrate reduction therapy for Gaucher disease.
National Gaucher Foundation. (n.d.). Substrate Reduction Therapy.
Shin, S-H., et al. (2007). Pharmacological chaperone corrects lysosomal storage in Fabry disease caused by trafficking-incompetent variants. American Journal of Physiology-Cell Physiology, 292(5), C1879-C1887.
Fabry Disease News. (2024, April 19). Chaperone therapy for Fabry disease.
National MPS Society. (n.d.). HSCT.
La-Fauci, G., & A.H. Schuchman. (2016). Gene Therapy for Lysosomal Storage Disorders: Recent Advances and Limitations. Journal of Inborn Errors of Metabolism and Screening, 4, 1-7.
Iimori, T., et al. (2023). Gene therapy for lysosomal storage diseases: Current clinical trial prospects. Frontiers in Genetics, 14, 1064924.
Platt, F. M., d'Azzo, A., Davidson, B. L., Neufeld, E. F., & Tifft, C. J. (2018). Lysosomal storage diseases. Nature Reviews Disease Primers, 4(1), 27.