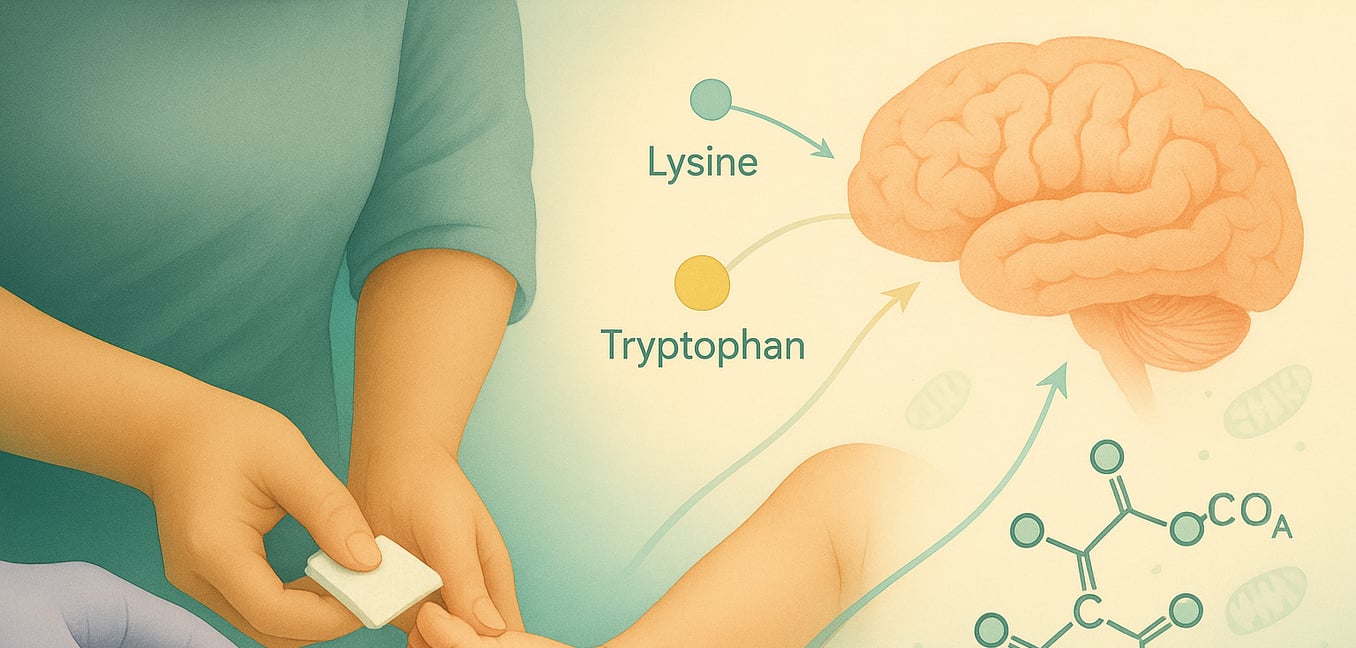



Glutaryl-CoA Dehydrogenase Deficiency (GA1), also known as glutaric acidemia type 1, is an inherited metabolic disorder. It stems from a deficiency in the glutaryl-CoA dehydrogenase (GCDH) enzyme, which is vital for breaking down the amino acids lysine, hydroxylysine, and tryptophan. This enzymatic defect leads to the harmful accumulation of substances like glutaric acid and 3-hydroxyglutaric acid, particularly damaging to the brain, especially the basal ganglia. GA1 is an autosomal recessive condition, meaning an affected child inherits one non-working copy of the GCDH gene from each parent. Understanding the diagnostic pathway is crucial for early intervention and management.
Newborn Screening: The First Line of Detection
Newborn screening programs play a pivotal role in the early identification of GA1, often before symptoms manifest. This early detection is key to preventing severe neurological damage.
- Screening Process: A small blood sample, obtained via a heel prick within days of birth, is analyzed. Laboratories test these dried blood spots for elevated C5-DC acylcarnitine (glutarylcarnitine), a primary marker indicating potential GA1.
- Interpreting Results: An out-of-range screening result is not a definitive diagnosis. It signals the need for further, more specific confirmatory tests to establish if GA1 is present.
- Importance of Early Detection: Identifying GA1 through newborn screening allows for immediate initiation of management strategies. This includes a specialized diet low in lysine and carnitine supplementation, significantly reducing the risk of encephalopathic crises and long-term neurological damage.
Biochemical Analyses: Confirming the Diagnosis
Following an abnormal newborn screen or if GA1 is suspected due to clinical symptoms, specific biochemical tests are performed to confirm the diagnosis by looking for characteristic metabolic signatures.
- Urine Organic Acid Analysis: This test measures levels of organic acids in the urine. In GA1, significantly increased concentrations of glutaric acid and 3-hydroxyglutaric acid are hallmark findings, resulting from the blocked metabolic pathway.
- Plasma Acylcarnitine Analysis: A blood plasma sample is analyzed for acylcarnitines. Elevated glutarylcarnitine (C5DC) is typically observed. However, C5DC levels can sometimes be normal or only slightly elevated if the body's carnitine stores are depleted, making urine analysis particularly important.
- Direct GCDH Enzyme Activity Measurement: This test directly assesses the function of the GCDH enzyme. It is usually performed on cultured skin cells (fibroblasts) or white blood cells (leukocytes). Markedly reduced or absent enzyme activity provides direct biochemical evidence of GA1.
Molecular Genetic Testing and Enzymatic Assays: Definitive Confirmation
Molecular genetic testing and enzymatic assays provide the most definitive confirmation of GA1 by examining the underlying genetic defect and its direct impact on enzyme function.
- GCDH Gene Analysis: This involves sequencing the GCDH gene to identify disease-causing variants (mutations). Over 200 such variants are known. Identifying two pathogenic variants, one inherited from each parent, confirms the diagnosis at a molecular level.
- Enzyme Activity Assays: As mentioned in biochemical analyses, these assays directly measure the GCDH enzyme's ability to process its substrate in patient-derived cells. A significant reduction in activity confirms the metabolic defect. While the level of residual enzyme activity can vary, it does not always directly correlate with clinical severity.
Neuroimaging Findings in GA1
Neuroimaging techniques, primarily Magnetic Resonance Imaging (MRI), are valuable for assessing brain involvement, especially if GA1 is diagnosed late or after an encephalopathic crisis. While not a primary diagnostic tool for initial identification, MRI can reveal characteristic changes. These often include:
- Frontotemporal atrophy (shrinkage of brain tissue in the frontal and temporal lobes).
- Widening of the Sylvian fissures, sometimes described as a "bat-wing" appearance due to incomplete opercularization.
- Abnormalities in the basal ganglia, such as increased signal intensity on T2-weighted images, indicative of damage from metabolite accumulation. These findings help understand the extent of neurological impact and can support the diagnosis in conjunction with biochemical and genetic results.
Prenatal Diagnosis and Genetic Counseling
For families with a previously affected child and known GCDH gene mutations, prenatal diagnosis offers options for future pregnancies. Genetic counseling is essential in these situations.
- Purpose: Prenatal diagnosis aims to determine if a developing fetus has GA1, allowing parents to make informed decisions and prepare for specialized care from birth if needed.
- Genetic Prenatal Testing: This involves analyzing fetal DNA obtained through chorionic villus sampling (CVS) around 10-13 weeks of gestation or amniocentesis around 15-20 weeks. The fetal DNA is tested for the specific GCDH mutations identified in the family.
- Biochemical Prenatal Testing: Metabolite levels, such as glutaric acid, 3-hydroxyglutaric acid, and glutarylcarnitine, can be measured in amniotic fluid obtained via amniocentesis. Elevated levels suggest the fetus is affected and can complement genetic testing.
- Genetic Counseling: Provides families with information about GA1, inheritance patterns, recurrence risks, and available testing options. It also supports carrier testing for other family members to understand their risk of having children with GA1.
