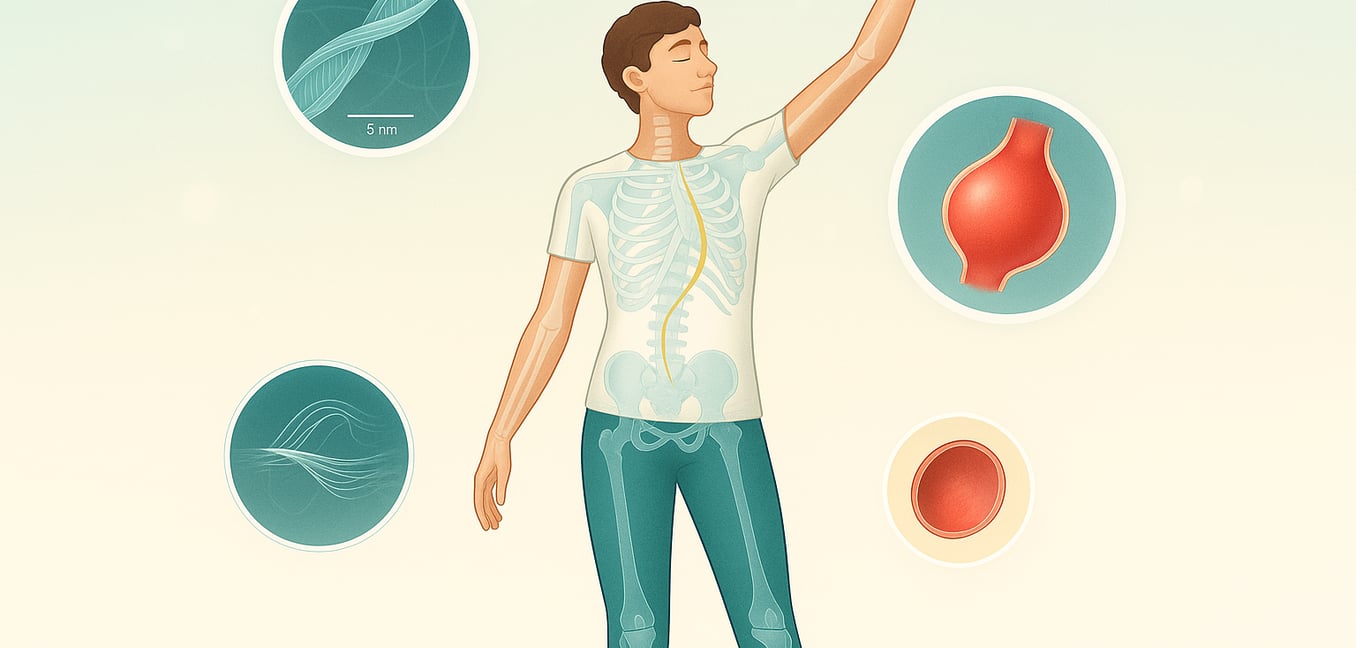



Marfan syndrome is a genetic disorder that affects the body's connective tissue, the strong, flexible material that provides structure and support to cells throughout the body. Caused by variants in the FBN1 gene, which provides instructions for making a protein called fibrillin-1, this condition weakens the tissues that are rich in this protein. Because connective tissue is a fundamental component of many different organ systems, the condition can cause a wide range of health issues. The heart, blood vessels, skeleton, and eyes are most frequently involved, but the specific symptoms and their severity can vary dramatically from one person to another, making diagnosis and management a highly individualized process.
The effects of Marfan syndrome are often most apparent in the skeletal system, leading to a characteristic body type and specific bone-related issues. One of the classic signs is a tall, slender physique with disproportionately long arms, legs, and fingers, a condition known as arachnodactyly, or "spider-like" fingers. This is due to the overgrowth of the long bones, and affected individuals may also have little muscle tone (hypotonia) and minimal fat under the skin. Another common skeletal symptom is a deformity of the chest wall, where the overgrowth of ribs pushes the breastbone (sternum) either inward to create a sunken appearance (pectus excavatum) or outward, resulting in a protruding chest (pectus carinatum). Curvature of the spine, known as scoliosis, is also frequently seen. This side-to-side curve can range from mild to severe and may worsen rapidly during adolescent growth spurts, sometimes leading to back pain. These skeletal features, along with others like unusually flexible joints (hypermobility) and flat feet, are key indicators that a doctor will look for during a physical evaluation.
Beyond the visible skeletal signs, Marfan syndrome presents serious and potentially life-threatening symptoms affecting the cardiovascular system. A critical concern is the progressive widening, or aneurysm, of the aorta—the main artery carrying blood from the heart. Weakened connective tissue in the aortic wall makes it prone to stretching, and without treatment, it can tear (aortic dissection) or rupture, which is a medical emergency. This weakening can also lead to aortic regurgitation, where blood flows backward into the heart. Another common heart issue is mitral valve prolapse, where the valve between the heart's left chambers becomes floppy and doesn't close properly. This can cause mitral regurgitation and, in severe cases, may lead to chest pain, abnormal heart rhythms, or congestive heart failure. In the eyes, a hallmark symptom is the dislocation of one or both lenses (ectopia lentis), which occurs in approximately 60 percent of individuals. This displacement, along with severe nearsightedness (myopia), an increased risk for retinal detachment, early-onset cataracts, and glaucoma, can result in significant vision loss if not carefully monitored and managed by an eye specialist.
What do people with MarFans look like?
Individuals with Marfan syndrome often have a distinct physical appearance, characterized by a tall, slender build with disproportionately long arms, legs, fingers (arachnodactyly), and toes. Their skeletal structure may show specific signs, such as a chest that appears sunken (pectus excavatum) or protrudes outward (pectus carinatum), and an abnormal side-to-side curvature of the spine (scoliosis). Distinctive facial features can include a long, narrow face, deep-set eyes, a small jaw, flat cheekbones, and crowded teeth. It is important to note, however, that the physical signs vary greatly among individuals, and not everyone with the condition will display all of these traits or have them to the same degree.
What disease mimics Marfan?
Several heritable connective tissue disorders can mimic Marfan syndrome (MFS) due to significant clinical overlap in cardiovascular, skeletal, and cutaneous features. Loeys-Dietz syndrome (LDS) is a primary example, sharing characteristics like aortic root aneurysms, scoliosis, and pectus deformities. However, LDS is often distinguished by more aggressive and widespread arterial disease, unique craniofacial features such as hypertelorism or a bifid uvula, and the absence of ectopia lentis (dislocated lens), a key feature of MFS. Additionally, certain types of Ehlers-Danlos syndrome (EDS) can present with similar findings; for instance, Kyphoscoliotic EDS may feature a "marfanoid habitus," and Hypermobile EDS can be associated with aortic root dilatation. The MASS (mitral, aortic, skin, and skeletal) phenotype is also considered a related but milder condition on the MFS spectrum.
At what age do you notice Marfan syndrome?
Although people are born with Marfan syndrome, its signs and symptoms can become noticeable at any age, from infancy through adulthood. The features of the condition, such as a tall, slender build and disproportionately long limbs, often become more apparent during the growth spurts of childhood and adolescence. As a result, the age of diagnosis varies significantly; research shows the median age for diagnosis is around 19 years, but many individuals are not identified until well into adulthood. Because the clinical signs can be subtle and develop over time, some people are said to "grow into the diagnosis" as they get older and the features become more distinct.
What are the facial features of a person with Marfan syndrome?
People with Marfan syndrome can have several distinct facial characteristics that contribute to a recognizable appearance. Their skull may be long and narrow ( dolichocephaly ), with other features including deep-set eyes ( enophthalmos ), flat cheekbones ( malar hypoplasia ), and a downward slant to the eyes. It is also common for individuals to have a small jaw ( micrognathia ) that may be recessed ( retrognathia ). Inside the mouth, a highly arched palate and crowded teeth are frequently observed, which can sometimes lead to issues with how the upper and lower teeth align when biting. While not every person will have all of these features, and their severity can vary greatly, this combination is characteristic of the condition.
What teeth problems do people with Marfan syndrome have?
Individuals with Marfan syndrome often experience a range of dental and oral issues stemming from the disorder's effect on connective tissue. A very common characteristic is a high, arched palate, which frequently leads to severe crowding of the teeth and a misaligned bite (malocclusion), such as a posterior crossbite. The teeth themselves may be unusually long and narrow, with potential root deformities or structural defects in the enamel and dentin. Internally, there is a higher prevalence of pulp calcifications, or "pulp stones," within the teeth. These structural challenges, particularly the crowding, can make effective oral hygiene difficult, which contributes to a greater susceptibility to periodontal diseases like gingivitis and periodontitis.
What is the finger test for Marfan syndrome?
The finger tests for Marfan syndrome are simple physical maneuvers used to check for disproportionately long fingers (arachnodactyly). The first is the thumb sign (or Steinberg sign), where you make a fist with your thumb tucked inside; a positive sign occurs if the tip of the thumb extends beyond the edge of your hand. The second is the wrist sign (or Walker-Murdoch sign), where you wrap your thumb and little finger around your other wrist; it's considered positive if your thumb and pinky overlap. These signs suggest the long, slender bone structure characteristic of the condition. While not definitive on their own, these findings are key indicators used by clinicians during a physical evaluation for Marfan syndrome.
How to differentiate between Ehlers-Danlos and Marfan syndrome?
Differentiating between Marfan syndrome and Ehlers-Danlos syndrome (EDS) relies on their distinct primary features and underlying genetic causes. Marfan syndrome, caused by a mutation in the FBN1 gene, is classically defined by a combination of skeletal issues (tall stature, long limbs), cardiovascular problems (especially aortic root aneurysm), and a highly specific eye condition called ectopia lentis (dislocated lens), which is not a feature of EDS. In contrast, EDS is a group of disorders affecting collagen, with hallmark signs being significant joint hypermobility, skin hyperextensibility, and fragile tissues that lead to atrophic scarring. While both conditions can have overlapping features like joint hypermobility and aortic issues, the presence of ectopia lentis strongly points to Marfan syndrome, whereas extreme skin fragility points toward EDS. Genetic testing is often used to confirm a diagnosis and distinguish between the two.
