



与罕见疾病的旅程往往从寻找答案开始。对于那些在溶酶体储存疾病(LSD)中导航的个人和家庭,这一搜索将你带入到我们自身细胞的微观世界。我们知道,这可能是一个令人生畏的领域,充满了复杂的科学术语和深刻的不确定性。我们的目标是与您一起走这条道路,将科学分解为清晰、支持性的信息。我们相信理解的力量,我们的承诺是提供一个空间,让您找到清晰、社区和 reassurance,您在这段旅程中并不孤单。
什么是溶酶体储存疾病?

从本质上讲,溶酶体储存疾病(LSD)是细胞的“循环中心”中的一种挑战,细胞的关键“回收中心,” 一个叫做溶酶体的细胞器。LSD的故事开始于 比利时科学家Christian de Duve的开创性工作,他在1950年代发现了溶酶体——这一成就让他在1974年获得诺贝尔奖。他指出这些微小的细胞器作为细胞的消化系统,充满了叫做酶的专门蛋白质。在健康的细胞中,溶酶体充满了可以把复杂材料(如脂肪和糖)分解成细胞可以重复使用的简单成分的酶(Platt, 2018)。
对于LSD患者来说,特定的基因突变意味着这些关键酶中的一种要么缺失,要么无法正常工作。大多数LSD是 常染色体隐性,这意味着一个孩子必须从父母双方遗传突变基因的一个副本才能患上这种疾病(Meikle et al., 1999)。
想象一下一个城市的回收系统,其中负责处理塑料的团队突然停止工作。塑料将开始堆积,导致城市各处出现问题。这与LSD中细胞内部发生的情况相似。特定材料(称为底物)开始积累,这正是缺失的酶本应加工的材料(Platt, 2018)。随着时间的推移,这种堆积使得溶酶体充满了储存的材料,这会损害细胞。这种“细胞垃圾危机”最终可能导致影响全身各个器官的一系列症状(Parenti, Andria, & Ballabio, 2015)。
诊断之旅:多样化的症状光谱

研究人员现在已经确定了超过 70种不同类型的LSD。虽然它们都源自溶酶体内的问题,但它们以多种方式表现出来,每种症状都由缺失的特定酶和随之积聚的底物定义。一些更广为人知的LSD包括:
高雪病: 由酶葡萄糖脑苷酶缺乏引起,导致称为葡萄糖脑苷的脂肪物质积聚(Grabowski & Hopkin, 2022)。
庞贝病: 涉及复杂糖类糖原的积累,尤其影响肌肉细胞,因为缺乏酶酸α-葡萄糖苷酶(Kishnani et al., 2006)。
法布瑞病: 由于缺乏α-半乳糖苷酶A,导致称为球三糖酰基氨基脂的脂肪在全身细胞中积聚(Germain, 2010)。
黏多糖贮积症(MPS): 一组相关疾病(包括Hurler、Hunter和Sanfilippo综合症),需要不同酶来分解称为糖胺聚糖(GAGs)的复杂糖分子(Parenti, Andria, & Ballabio, 2015)。
尼曼-皮克病(A、B、C型)& 泰-萨克斯病: 其他知名LSD,影响身体代谢脂肪(脂质)的能力,导致严重的神经损伤(Platt, 2018)。
因为溶酶体在几乎每个细胞中都存在,所以LSD的症状极为多样且逐渐加重。它们可以影响大脑和神经系统、心脏、骨骼系统、肝脏、脾脏、肺,甚至眼睛(Parenti, Andria, & Ballabio, 2015)。我们知道每段旅程都是不同的,即使在同名情况下,病情的严重程度和发病年龄也可能具有显著差异。
导航寻找答案的道路
对于许多家庭来说,诊断的道路是漫长而艰辛的过程,这一经历通常被称为“诊断之旅”(The LSD Collaborative, 2015)。早期症状可能微弱、不明确,可能模仿更常见的儿童疾病,导致令人沮丧的延误和误诊(Wraith, 2002)。我们知道这种不确定的时期是多么艰难,这突显出在未专注于这些罕见疾病的人群中,提高意识的重要挑战。一项 研究 显示,接近89%的受访初级保健医生甚至从未将LSD作为其患者任何潜在诊断的考虑,这突显了 awareness 的重要挑战。
终于得到诊断的解脱感——对令人困惑和痛苦症状有了名称——可以是巨大的。一个名叫P.的患者分享道,她的症状始于童年,但直到数十年后才被诊断出,在此期间,她的家人已经遭受了严重的痛苦。
诊断方法已变得日益复杂。它们通常涉及:
酶分析: 血液测试可以测量特定溶酶体酶的活性水平。显著降低或缺失的水平是LSD的关键指标(Sun, 2018)。
基因测试: 通过识别负责制造酶的基因中的具体突变来确认诊断,这有时可以帮助预测疾病的严重程度或指导治疗选择(Parenti, Andria, & Ballabio, 2015)。
新生儿筛查(NBS): 近年来,越来越多的地区将可治疗的LSD,如庞贝病、高雪病、法布瑞病和MPS I,添加到他们的标准新生儿筛查面板中(Burton, 2017)。通过在生命的头几天识别这些病症,在重大和不可逆转的损害发生之前,NBS提供了改变疾病进程的最佳机会。
人类的代价:不仅仅是医疗档案
我们知道,LSD的影响远超身体症状。这些是慢性、渐进式且往往使人心力交瘁的疾病,给患者及其家庭带来了深刻的医学、情感、社会和经济负担。日常现实可能包括管理慢性疼痛和令人虚弱的疲惫。一个名叫Ted的人患有 高雪病,形容他经历着的疲惫是一种完全不同于普通疲劳的不可动摇的疲惫,使即使是简单的任务也变得艰巨。
情感的画面复杂,充满了管理慢性疾病的压力、对未来的不确定以及伴随健康下降的悲伤。正如Sophia的母亲Jennie分享的那样,她的女儿患有 庞贝病,在诊断后的时期是可怕的:“最可怕的部分……就是没有确切答案,不知道会发生什么或何时发生。” 这是一个重塑家庭动态和生活选择的旅程,我们致力于提供一个社区,让这些深刻的个人经历可以得到分享、确认和理解。
一个想法变为现实
几十年来,这些疾病的治疗选择悲惨地有限。但是,1964年由溶酶体的发现者Christian de Duve首次构思的革命性想法改变了一切(Beck, 2018)。这个概念简单而深刻:如果我们能够从外部替换缺失的酶呢?这一洞察为酶替代疗法(ERT)奠定了基础(de Duve, 1964)。
从概念到临床的旅程充满了巨大的科学奉献。1970年代,关键实验表明,实验室培养皿中的缺乏细胞可以吸收健康细胞分泌的缺失酶,这一现象称为“交叉校正。”(Fratantoni, Hall, & Neufeld, 1968)。这证明了基本原理可以工作。进一步的研究发现了能指导酶到达溶酶体的细胞“地址标签”:一种叫做甘露糖-6-磷酸(M6P)的糖分子,可以与细胞表面上特定的M6P受体结合(Kornfeld, 1986)。
真正的临床突破是由 Roscoe Brady博士及其团队在国家卫生研究院(NIH)主导的(Beck, 2018)。他们专注于高雪病,从人类胎盘中提纯所需的酶(葡萄糖脑苷酶),并通过生化改造使其能够靶向合适的细胞(Brady et al., 1974)。临床试验的成功令人震惊,导致了第一个FDA批准的ERT,Ceredase™,在1991年获批(Beck, 2018)。这标志着一个新时代的开始。
如今,感谢重组DNA技术,ERT不再是源自人类组织。它们在安全的实验室环境中以大量安全和一致的方式生产,这是一个重大进步,提高了安全性和供应(Platt, 2018)。
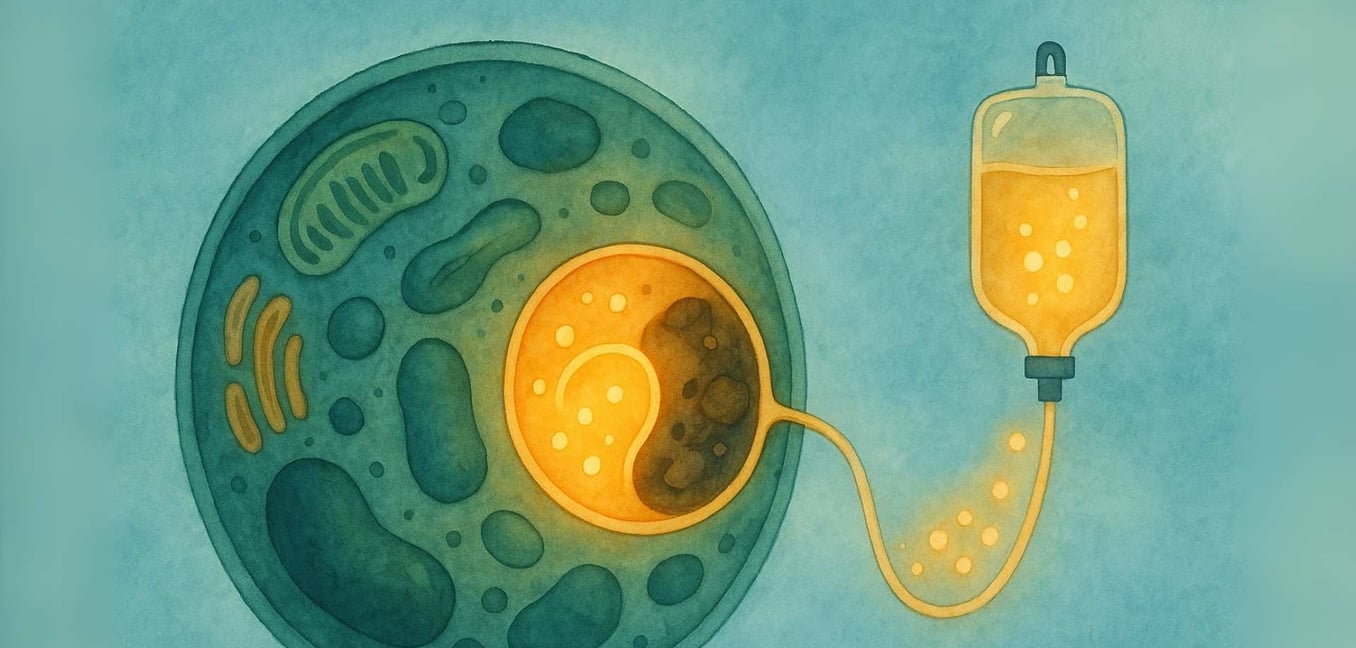
酶替代疗法(ERT)是如何工作的

ERT是一种医疗治疗,旨在将功能性的缺失酶直接传递到最需要它的细胞。以下是该过程的详细介绍:
输注: 该疗法通常以静脉注射(IV)方式施用,通常在几小时内缓慢输注,每一到两周进行一次(Beck, 2018)。
传递系统: 这些酶经过工程处理,带有特别的M6P“地址标签”,使得身体的细胞可以识别。这使得酶能够与细胞表面的M6P受体结合,像钥匙插入特定锁那样(Kornfeld, 1986)。
细胞作用: 一旦细胞识别并与酶结合,它就会通过一种称为内吞作用的过程将其引入内部,并直接引导到溶酶体。在那里,替换酶最终可以开始工作,分解积聚的材料,帮助恢复细胞内的更健康平衡(Kornfeld, 1986)。
治疗旅程:希望与诚实的期望
ERT是一种治疗,而非治愈(Platt, 2018)。它并不纠正潜在的基因缺陷,需要终生定期输注的承诺,这是患者及其家庭生活的重要部分(Beck, 2018)。
此外,ERT面临几项重大挑战:
血脑屏障(BBB): 这是无疑最大的挑战。血脑屏障是一个保护盾,防止大分子(包括ERT酶)进入大脑。由于估计三分之二的LSD都有神经症状,标准的ERT往往无法解决认知下降或其他中枢神经系统问题,即使它对身体的其他部分有效。
免疫反应: 由于源自蛋白质的输注酶,身体有时会将其视为“外来物”,并产生抗药物抗体(ADA)。这些抗体在某些情况下可能会降低疗法的有效性,或导致与输注相关的反应(IAR),如发热、皮疹,或更严重的过敏反应。
递送至“避难点”: 除大脑之外,某些组织对大分子酶的到达是困难的。这些包括致密骨、无血管软骨和心脏瓣膜,意味着尽管接受治疗,骨骼问题和瓣膜性心脏病可能会持续进展。
扩展治疗武器库

ERT的局限性激励了研究人员和我们的社区不断推动新疗法的研发。未来的治疗集中在对这些疾病的多管齐下的攻击上,几种令人兴奋的策略正在涌现:
底物减少疗法(SRT): 此方法不使用酶替代,而是利用口服药物抑制最初生成底物所需的酶。这有效地“减轻了”受损溶酶体的负担。已批准的药物如 Miglustat 和 Eliglustat 用于一型高雪病。
药理性伴侣疗法(PCT): 对于那些体内产生错误折叠但仍具一定功能的酶的患者,这些小型口服药物充当“支架”。它们与不稳定的酶结合,帮助其正确折叠,从而通过细胞的质量控制,并到达溶酶体发挥作用。Migalastat是针对特定“可合适”突变的法布瑞患者的批准伴侣。
造血干细胞移植(HSCT): 对于某些具有中枢神经系统累及的严重LSD,如MPS I(Hurler综合症),来自健康供体的HSCT(或骨髓移植)可以是一种选择。其想法是供体来源的细胞将到达大脑,并提供缺失酶的局部、持续来源。然而,这是一个风险极高的程序,具有重大潜在并发症。
基因疗法: 这是一个最令人激动且潜在变革的前沿领域。目标是将功能性基因的拷贝导入患者自己的细胞,使其能够自行产生酶。使用工程病毒载体或其他递送系统,研究人员正在探索提供持久的、潜在一次性治疗的方法,以纠正疾病的根本原因。对于庞贝病、法布瑞病和几种MPS疾病,目前正在进行临床试验,提供了强大的希望源泉。
共同进步,携手前行
从溶酶体的基础发现到ERT的开发以及基于基因医学的曙光,见证了科学探究和人类韧性的力量。我们致力于继续这一前进的步伐,一起携手前进。我们将始终努力提供可信、清晰的信息,培养一个支持和关爱的社区,并倡导带来实际希望的研究,为更光明的未来铺平道路。
我们邀请您收听最新的播客节目,简明扼要地概述酶替代疗法。它旨在将这个复杂主题分解为易于理解的部分。
<iframe width="560" height="315" src="https://www.youtube.com/embed/T91yim2x8ac?si=5aRF203Rw9fqjAoN" title="YouTube视频播放器" frameborder="0" allow="accelerometer; autoplay; clipboard-write; encrypted-media; gyroscope; picture-in-picture; web-share" referrerpolicy="strict-origin-when-cross-origin" allowfullscreen></iframe>
参考文献
诺贝尔奖外展有限公司。(2024)。1974年诺贝尔生理学或医学奖。NobelPrize.org。
de Duve, C.(2013)。Christian de Duve:通过使用离心机发现新细胞器的细胞探险家。《国家科学院院刊》,110(31),12559-12561。
洛克菲勒大学。(n.d.)。“用离心机探索细胞”:溶酶体的发现。医院百年庆。
MSD手册专业版。(n.d.)溶酶体储存疾病概述。
哥伦比亚大学欧文医学中心。(n.d.)儿童溶酶体储存疾病。
克利夫兰诊所。(2022年6月27日)。溶酶体储存疾病和障碍。
克利夫兰诊所。(2022年6月27日)。溶酶体储存疾病和障碍。
生物科学研究所。(n.d.)溶酶体储存疾病(LSDS)。
van der Meijden, J. C.等(2010)。“谷歌医生”终结溶酶体储存疾病的诊断之旅。《儿童疾病档案》,95(8),642-644。
Medicover遗传学。(2025年2月26日)。诊断之旅:寻找罕见疾病答案。
南非罕见疾病组织。(2023年2月28日)。应对溶酶体储存疾病诊断和治疗的挑战。
格林伍德基因中心。(2024年8月14日)。溶酶体储存疾病:高雪病和法布瑞病。
维基百科。(n.d.)酶替代疗法。
Beck, M.(2018)。酶替代疗法及其以外的领域——悼念Roscoe O. Brady, M.D.(1923-2016)。《遗传代谢疾病杂志》,41(1),3-13。
Park, J. J., & Lee, K.(2022)。甘露糖-6-磷酸糖苷用于溶酶体靶向:从酶替代疗法到靶向溶酶体的嵌合体的各种应用。《动物细胞与系统》,26(3),84-91。
美国食品和药物管理局(FDA)。(1991)。搜索孤儿药物指定和批准:Ceredase。
国家高雪病基金会。(n.d.)ERT批准25周年。
Begley, D. J.(2015)。改变血脑屏障运输,以给溶酶体储存病患者带来希望。《脑血流与代谢杂志》,35(1),3-5。
Pardridge, W. M.(2015)。靶向治疗性溶酶体酶的血脑屏障。《溶酶体储存疾病》,327-343。
Aflaki, E.等(2018)。酶替代疗法:最佳选择是什么?《当前药物设计》,24(11),1238-1250。
Giugliani, R.等(2018)。针对黏多糖贮积症的酶替代疗法:新的发展和临床结果。《孤儿药物专家意见》,6(4),277-287。
高雪病新闻。(n.d.)。针对高雪病的底物减少疗法。
国家高雪病基金会。(n.d.)。底物减少疗法。
Shin, S-H.等(2007)。药理伴侣纠正因转运无能变体而导致的法布瑞病溶酶体储存。《美国生理学杂志-细胞生理学》,292(5),C1879-C1887。
法布瑞病新闻。(2024年4月19日)。法布瑞病的伴侣疗法。
国家MPS协会。(n.d.)。HSCT。
La-Fauci, G., & A.H. Schuchman.(2016)。溶酶体储存疾病的基因治疗:近期进展与局限性。《先天代谢缺陷与筛查杂志》,4,1-7。
Iimori, T.等(2023)。溶酶体储存疾病的基因治疗:当前临床试验前景。《遗传学前沿》,14,1064924。
Platt, F. M., d'Azzo, A., Davidson, B. L., Neufeld, E. F., & Tifft, C. J.(2018)。溶酶体储存疾病。《自然评论-疾病前沿》,4(1),27。